
Anemia aplásica (AA)
1 ¿Qué es la AA?
2 Síntomas
3 Diagnóstico
4 Evolución clínica
5 Tratamiento
6 Pronóstico
7 Registro
Se trata de una traducción automática. Nuestros textos no sustituyen la consulta con un médico. Si es posible, consulte siempre a uno de nuestros especialistas.
1 ¿Qué es la AA?
1.1 Generalidades
La anemia aplásica es una enfermedad hematológica no maligna. Se debe a un trastorno de la función de la médula ósea que provoca una disminución de la producción de células sanguíneas.
Según la causa de la anemia aplásica, se distingue entre formas congénitas (por ejemplo, anemia de Diamond-Blackfan o anemia de Fanconi, telomeropatías) y formas adquiridas. Ambas formas pueden aparecer a cualquier edad.
Los trastornos de la formación de sangre tras una quimioterapia o radioterapia no se denominan anemia aplásica.
1.2 Incidencia (epidemiología)
La frecuencia de la enfermedad (incidencia) en Europa Central es de 2-3 nuevos casos por cada millón de personas al año. Por lo tanto, la anemia aplásica es una enfermedad muy rara. La mayoría de los afectados enferman entre los 10 y los 25 años, así como a partir de los 60 años, y afecta por igual a ambos sexos.
1.3 Origen (patogénesis)
Según algunos estudios, en la anemia aplásica adquirida, una parte del propio sistema inmunitario ataca a las células de la médula ósea. Se trata de un subtipo de linfocitos que, mediante este proceso autoinmune, impide la formación de nuevas células sanguíneas. En algunos casos de anemia aplásica adquirida, se sospecha que la causa son medicamentos o sustancias tóxicas (aproximadamente el 20 %) o una infección viral (aproximadamente el 5 %).
Sin embargo, en la mayoría de los casos de anemia aplásica adquirida (aproximadamente el 75 %), no se puede determinar la causa del desarrollo de la enfermedad, por lo que el origen de la enfermedad sigue sin estar claro (idiopático). Sin embargo, también hay casos en los que una alteración congénita es responsable de la aparición o el curso de la anemia aplásica.
Estudios recientes muestran que una parte relevante de los pacientes con anemia aplásica aparentemente adquirida padecen una forma congénita que solo se manifiesta clínicamente de forma tardía. Actualmente, la proporción de casos congénitos que solo se detectan en la edad adulta es del 5-15 % de todas las anemias aplásicas en adultos. Es de suponer que esta proporción seguirá aumentando gracias a la mejora de las posibilidades de diagnóstico. Esto es relevante, ya que la terapia recomendada para este grupo de pacientes difiere de la de los casos de anemia aplásica adquirida. Esto es especialmente importante en lo que respecta al trasplante de células madre.
1.4 Criterios de diagnóstico y clasificación
Para poder diagnosticar una anemia aplásica, deben cumplirse los siguientes criterios:
- El número de células en la médula ósea (celularidad) es inferior al 25 % en comparación con la médula ósea sana, lo que se evalúa mediante una biopsia de médula ósea. La producción de células puede verse reducida (hipoplásica) o desaparecer por completo (aplásica) como consecuencia de la enfermedad.
- Disminución de dos (bicitopenia) o tres series celulares (tricitopenia o pancitopenia) en diferentes grados en el frotis sanguíneo.
- No hay indicios de formación (nueva) de tejido conjuntivo en la médula ósea (fibrosis) ni de afectación de la médula ósea por células malignas o ajenas a la médula ósea.
- Además, no debe haber alteraciones celulares significativas (displasias) en la formación de la sangre (hematopoyesis).
- No se ha realizado recientemente ninguna radioterapia o quimioterapia que pueda explicar una alteración de la función de la médula ósea (insuficiencia medular).
- No ha habido contacto con radiación radioactiva.
La anemia aplásica se clasifica en función de los valores sanguíneos (véase la tabla siguiente) en:
- No grave Anemia aplásica = nSAA («non-severe AA»)/Anemia aplásica moderada («Moderate AA»)
- Anemia aplásica grave = SAA («severe AA»)
- Anemia aplásica muy grave = vSAA («very severe AA»)
y es de vital importancia para el pronóstico y el tratamiento.
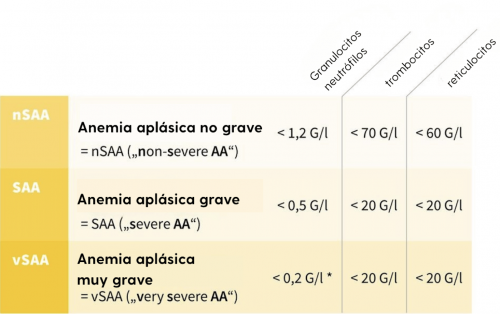
Clasificación de la anemia aplásica según el hemograma (recuento celular y frotis). Deben cumplirse dos de los tres criterios. *Para la clasificación como vSAA, es obligatorio cumplir el criterio de granulocitos < 0,2 G/l.
2 Síntomas
2.1 Anemia
Una disminución de los glóbulos rojos (eritrocitos) que transportan oxígeno puede provocar debilidad, cansancio y dificultad para respirar, e incluso taquicardia, especialmente durante el esfuerzo físico. Además, los pacientes con anemia suelen presentar palidez, especialmente en las palmas de las manos, aunque la presencia de palidez no debe interpretarse como prueba de anemia.
2.2 Mayor susceptibilidad a las infecciones
La disminución del número de glóbulos blancos (leucocitos) aumenta la susceptibilidad del organismo a las infecciones. Dado que el sistema inmunitario del organismo no funciona adecuadamente cuando el número de neutrófilos, un subtipo de glóbulos blancos, es bajo, una infección de este tipo puede convertirse en una amenaza para la vida en cuestión de horas y provocar una septicemia.
Por lo tanto, es importante que informe inmediatamente a su médico si presenta fiebre. Se considera fiebre una temperatura corporal superior a 38 °C medida dos veces en una hora en el oído o superior a 38,3 °C medida una vez en el oído.
2.3 Hemorragias
Cuando el número de plaquetas (trombocitos) es bajo, la coagulación sanguínea puede verse alterada. Esto provoca sangrado de las encías y la aparición de petequias, pequeñas hemorragias puntiformes en la piel, o hematomas. Estos pueden aparecer de forma espontánea, es decir, sin que haya habido una lesión previa. Cuando la coagulación sanguínea se ve alterada, incluso una hemorragia o lesión relativamente leve (por ejemplo, durante una visita al dentista) puede convertirse en una amenaza. Por lo tanto, en caso de hemorragia, debe ponerse en contacto con su médico lo antes posible para que este pueda decidir si es necesario tomar medidas especiales (por ejemplo, una transfusión de plaquetas).
3 Diagnóstico
Si se presenta uno o varios de los trastornos y síntomas mencionados anteriormente, el médico de cabecera solicitará un análisis de sangre. Si se detecta alguna irregularidad en el hemograma, se derivará al paciente a un especialista en hematología y oncología.
Allí se realizarán una serie de pruebas adicionales:
- Historial médico (anamnesis), también del familiar, incluyendo un registro detallado de los medicamentos tomados.
- Exploración física, p. ej., signos de anemia y hemorragia.
- Exámenes celulares
- Hemograma diferencial microscópico
- Reticulocitos
- Diagnóstico de HPN (hemoglobinuria paroxística nocturna) (se detecta un número relevante de células HPN en hasta el 70 % de los casos de AA), véase HPN, 3 Diagnóstico
- Química clínica
- Parámetros de hemólisis: en particular, LDH, haptoglobina, bilirrubina
- Coagulación: valor de Quick, PTT, fibrinógeno
- Parámetros de la función hepática: AST, ALT y AP
- Parámetros de la función renal: creatinina, ácido úrico
- Glucosa en sangre
- Proteínas totales, electroforesis, inmunoglobulinas
- Parámetros inflamatorios CRP
- Niveles de vitamina B12 y ácido fólico
- Estado del hierro: ferritina. Si los valores de ferritina son > 1000 ng/ml, se debe investigar más a fondo la posible daño orgánico causado por una posible sobrecarga de hierro
- Diagnóstico de virus: hepatitis A, B, C; VIH, VEB, CMV, parvovirus B19
- Anticuerpos antinucleares y anti-ADN
- Diagnóstico funcional
- Ecografía cardíaca y de la parte superior del abdomen (sonografía)
- Radiografía de tórax
- ECG
- Pruebas especiales
- Tipificación HLA del paciente y sus hermanos
- Determinación de la longitud de los telómeros
- En caso de sospecha de síndrome de insuficiencia medular «congénita», diagnóstico adicional, p. ej., análisis de rotura cromosómica en caso de sospecha de anemia de Fanconi, pruebas genéticas
Si se confirma una disminución del número de una o varias series de células sanguíneas sin que se conozca la causa del aumento del consumo o la degradación de estas células sanguíneas, se debe realizar urgentemente un examen de la médula ósea. De esta manera se puede determinar si existe un trastorno de la formación de la sangre u otra causa.
Para ello se realiza una punción de médula ósea, que puede realizarse de forma ambulatoria. Bajo anestesia local, se extrae un cilindro óseo del paciente, generalmente del hueso pélvico, con una aguja hueca (aguja de Jamshidi) (biopsia de médula ósea, punción de médula ósea). Este tiene una longitud aproximada de 1,5 cm y un diámetro de 2-3 mm, y se examina y evalúa al microscopio (histología).
Además, se extraen muestras de sangre, médula ósea y médula grasa («fragmentos de médula») (aspiración de médula ósea), que se extienden en un portaobjetos, se secan, se tiñen y también se evalúan con el microscopio en su totalidad o en su posición relativa (examen citológico).
Además, se pueden realizar exámenes genéticos en las células de la médula ósea, cuyos resultados pueden facilitar, por ejemplo, la diferenciación de otras enfermedades.
Dado que los distintos pasos de laboratorio para la preparación de la histología de la médula ósea requieren mucho tiempo, se necesitan entre una y dos semanas para obtener un resultado completo. Si se observa una disminución en la formación de dos o tres filas de células (eritrocitos, leucocitos, trombocitos) de acuerdo con los criterios diagnósticos sin la presencia simultánea de células patológicamente alteradas (por ejemplo, células leucémicas) y sin que haya habido previamente quimioterapia o radioterapia, se habla de anemia aplásica.
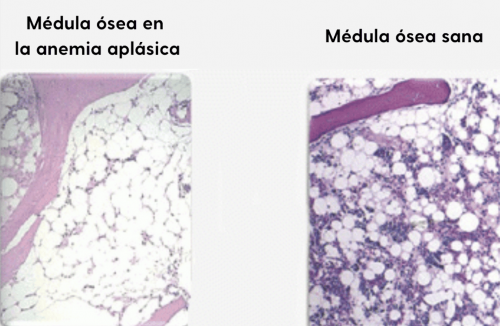
Hallazgo en la médula ósea de un paciente con anemia aplásica en comparación con la médula ósea sana. En la médula ósea enferma se reconocen principalmente tejido conjuntivo y células grasas. En la médula sana, las células sanguíneas se distinguen de las grandes células grasas blancas como pequeños puntos de diferentes colores.
El objetivo de estas numerosas pruebas es
- descartar otras enfermedades,
- aclarar las posibles causas (etiología),
- determinar la gravedad de la anemia aplásica
- y establecer el pronóstico.
En pacientes con anemia aplásica muy grave y grave, menores de 50 años y en buen estado físico, es conveniente realizar una tipificación HLA del paciente directamente en el momento del diagnóstico. Si hay hermanos, también deben ser tipificados para determinar su idoneidad para la donación de células madre.
4 Evolución
Sin un tratamiento específico, la anemia aplásica en la edad adulta es mortal en hasta el 70 % de los casos.
Existe la posibilidad de que la anemia aplásica derive en un síndrome mielodisplásico (SMD) o en una leucemia mieloide aguda (LMA). Además, algunos pacientes con AA presentan una mutación específica de la HPN, por lo que también pueden ser relevantes los síntomas y la necesidad de tratamiento de la hemoglobinuria paroxística nocturna (HPN).
5 Tratamiento
5.1 Resumen
Las curaciones hematológicas espontáneas (remisiones espontáneas) prácticamente no se dan en casos de insuficiencia medular grave.
El tratamiento es necesario en los siguientes casos
- anemia aplásica muy grave (vSAA) y grave (SAA)
- anemia aplásica no grave (nSAA) con una disminución significativa de al menos una línea celular (citopenia), que requiere transfusiones regulares o conlleva un riesgo de infección o hemorragia
Mientras que hace unas décadas apenas había perspectivas de curación o mejora a largo plazo, hoy en día existen opciones prometedoras. Para el tratamiento se dispone principalmente de dos medidas terapéuticas: la denominada terapia inmunosupresora (IST) y el trasplante de células madre (SZT) o trasplante de médula ósea (KMT). Además, existen terapias especiales para determinados subgrupos de pacientes. La elección de la terapia adecuada para un paciente concreto depende de la gravedad de la enfermedad, la edad y las posibles enfermedades concomitantes del paciente, así como del grado de compatibilidad HLA con un posible donante de médula ósea, ya sea familiar o no.
Si existe una indicación terapéutica, el tratamiento debe iniciarse lo antes posible para evitar la progresión de la enfermedad y sus posibles complicaciones (por ejemplo, anemia grave, infecciones, hemorragias y trastornos de la coagulación). Por lo tanto, es importante planificar el tratamiento de forma temprana en colaboración con un centro especializado.
El proceso terapéutico para pacientes con anemia aplásica muy grave y grave se muestra en la siguiente ilustración.
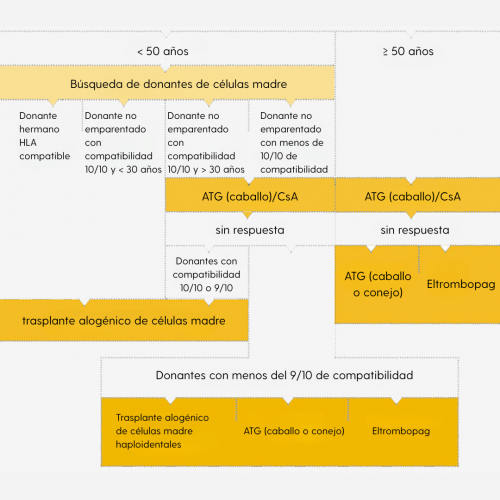
Representación claramente simplificada del algoritmo terapéutico. El algoritmo completo se encuentra en las directrices de la DGHO en Onkopedia.
5.2 Terapia inmunosupresora (IST)
Antitimocito globulina (ATG) y ciclosporina (CsA)
Dado que en la anemia aplásica adquirida el sistema inmunitario del propio organismo se vuelve contra la médula ósea, a menudo se recomienda un tratamiento inmunosupresor, especialmente en los siguientes casos:
- Pacientes con vSAA o SAA > 50 años
- Pacientes sin donantes HLA compatibles (hermanos)
- Pacientes con vSAA o SAA < 50 años en mal estado físico
- Pacientes con nSAA/MAA con riesgo de citopenia grave en al menos una línea celular
La terapia inmunosupresora suele consistir en una combinación de los medicamentos antitimocito globulina y ciclosporina. Esto permite que la médula ósea se recupere. Durante el transcurso de la terapia, suele producirse inicialmente un empeoramiento a corto plazo del hemograma, antes de que se produzca una mejoría.
La ATG es un anticuerpo que destruye los linfocitos T hiperactivos que dañan la médula ósea. Por lo general, la ATG se administra durante 4-5 días en forma de infusión en una vena grande a través de un catéter venoso central (ZVK). Durante el tratamiento con ATG, el recuento de plaquetas debe aumentarse o mantenerse en 30 G/l mediante transfusiones de plaquetas, ya que durante el tratamiento puede producirse una rápida disminución del recuento de plaquetas. Para el tratamiento con ATG hay que contar con una estancia hospitalaria de aproximadamente 1-2 semanas. Los efectos secundarios del tratamiento con ATG pueden ser reacciones alérgicas como erupciones cutáneas y fiebre. Para suprimir los efectos secundarios agudos del ATG, se administra además durante un breve periodo de tiempo un preparado de cortisona, por ejemplo, prednisona o prednisolona. Además, existe un mayor riesgo de contraer determinados agentes patógenos, por lo que deben tomarse las precauciones oportunas.
Otro factor esencial para la respuesta terapéutica de la enfermedad es la ciclosporina, que inhibe la liberación de sustancias inmunoestimulantes. Con la CsA se realizan controles de laboratorio periódicos para lograr el efecto óptimo, si es necesario, ajustando la dosis. El objetivo es alcanzar un nivel mínimo de 170-225 ng/ml en sangre. Para mantener un nivel estable, el medicamento debe tomarse con mucha regularidad, a intervalos fijos de 12 horas.
Los posibles efectos secundarios del tratamiento con CsA son infecciones, deterioro de la función renal, aumento de la presión arterial, hiperplasia gingival (hiperplasia gingival), aumento del crecimiento del vello, calambres musculares, trastornos sensoriales o temblores (tremor). Los efectos secundarios dependen de la dosis y suelen desaparecer al finalizar el tratamiento con CsA.
La CsA se toma en forma de cápsulas o jarabe durante al menos 12 meses. Si la respuesta al tratamiento es muy buena y estable, es importante reducir la dosis de CsA muy lentamente y de forma gradual para evitar una recaída de la enfermedad. Sin embargo, en algunos pacientes es necesario administrar CsA durante más tiempo o de forma permanente para mantener el éxito del tratamiento.
Gracias a la terapia inmunosupresora intensificada, en aproximadamente el 50-75 % de los pacientes se puede lograr la curación (remisión completa, RC) o, al menos, una mejora significativa (remisión parcial, RP), con independencia de las transfusiones y una reducción significativa del riesgo de infección y hemorragia. Se tarda entre 2 y 4 meses, y en algunos pacientes hasta 6 meses, en observar una mejora en los valores sanguíneos. El objetivo es eliminar los síntomas clínicos y los riesgos. Para ello no es necesaria una normalización completa de los valores sanguíneos, lo que a menudo no se consigue.
Si no se observa respuesta, se puede considerar la repetición del tratamiento inmunosupresor al cabo de 4-6 meses.
En caso de recaída (recurrencia), también es posible repetir el tratamiento inmunosupresor, ya que la probabilidad de una nueva respuesta es del 30-60 %.
Como complemento a un tratamiento específico, todos los pacientes deben recibir un tratamiento de apoyo.
Alemtuzumab
Existen otros medicamentos que actúan mediante el mismo mecanismo de inmunosupresión. Entre ellos se encuentra, por ejemplo, el alemtuzumab, un anticuerpo que actúa contra los linfocitos T. Este medicamento se utiliza en la leucemia linfocítica crónica (LLC) o la esclerosis múltiple (EM), pero también ha mostrado buenas tasas de respuesta en estudios sobre anemia aplásica, especialmente en pacientes de edad avanzada. Una ventaja de este medicamento es que solo se inyecta bajo la piel, por lo que no es necesaria la hospitalización. Si el paciente ha tenido anteriormente una infección por el virus citomegalovirus (CMV), se debe controlar regularmente este valor sanguíneo, ya que esta infección viral puede reaparecer durante el tratamiento.
Los pacientes en los que otros tratamientos no habían surtido efecto mostraron tasas de respuesta del 37-48 % con alemtuzumab.
5.3 Trasplante alogénico
En pacientes de hasta aproximadamente 50 años de edad (lo importante es el estado general, «la edad biológica») con anemia aplásica grave o muy grave (SAA o vSAA) y disponibilidad de un donante hermano que sea totalmente compatible con el paciente en las estructuras de compatibilidad tisular (HLA) (HLA idéntico), el tratamiento preferido (terapia de primera línea) es un trasplante alogénico.
Los pacientes menores de 30 años también pueden recibir células madre de un donante no emparentado HLA idéntico (donante externo) si no tienen un donante familiar HLA idéntico. Es importante que se realice una tipificación detallada (al menos 10 antígenos HLA) y que el donante y el receptor sean completamente idénticos en este aspecto.
En los últimos años, se ha reducido considerablemente la tasa de complicaciones en los trasplantes de donantes externos HLA compatibles. Por lo tanto, el trasplante de donante externo HLA idéntico se utiliza cada vez más en pacientes menores de 50 años que no han respondido al tratamiento inmunosupresor.
El objetivo del trasplante alogénico es sustituir la médula ósea no funcional del paciente por células madre sanas de un donante. Para ello, primero se destruye la médula ósea del paciente mediante diversas medidas (quimioterapia, terapia con anticuerpos, radiación). Este proceso, denominado «acondicionamiento», se lleva a cabo en los días previos al trasplante.
Paralelamente, se recogen células madre nuevas y sanas de un donante voluntario sano, ya sea familiar o no.
Las células madre se pueden obtener directamente de la médula ósea mediante una biopsia de médula ósea bajo anestesia. Las punciones en la cresta ilíaca para extraer la médula ósea pueden provocar hematomas y dolor que pueden durar varios días. Además, existe el riesgo general de la anestesia.
Como alternativa, se inyecta al donante durante varios días un medicamento que estimula la formación de granulocitos (G-CSF). Las células madre sanguíneas producidas de este modo migran desde la médula ósea a la sangre. Estas células madre sanguíneas periféricas (PBSZ) se extraen entonces con un dispositivo especial (aféresis), como en una donación de plasma sanguíneo. El procedimiento puede provocar molestias y dolores similares a los de la gripe.
Si las células madre se obtienen directamente de la médula ósea, se habla de un trasplante de médula ósea (TMO); si la obtención de células madre se realiza mediante aféresis, se denomina trasplante de células madre (TCM).
Los estudios indican que el tratamiento de la anemia aplásica con células madre de la sangre periférica puede ir acompañado de un aumento de las complicaciones, como reacciones de rechazo agudas o crónicas. Por lo tanto, siempre que sea posible, se deben utilizar células madre extraídas directamente de la médula ósea.
Independientemente de cómo se hayan obtenido las células madre, se purifican y se analizan para detectar agentes infecciosos. A continuación, el paciente recibe las células madre donadas. El trasplante en sí se realiza como una transfusión de sangre. Si todo va bien, las células madre del donante «crecen» y dan lugar a una función normal de la médula ósea y a la formación de sangre. Para un trasplante es necesaria una estancia hospitalaria de al menos 4 semanas.
Durante el trasplante, el paciente recibe medicamentos para prevenir (profilaxis) infecciones por bacterias y hongos. Además, se administran durante varios meses un preparado de cortisona, por ejemplo, prednisolona, y ciclosporina (CsA) para influir en el sistema inmunológico.
Las posibles complicaciones del trasplante son:
- Efectos secundarios tóxicos durante la terapia de acondicionamiento
- Infecciones
- Enfermedad de injerto contra huésped (EICH)
- Rechazo del trasplante
Enfermedad de injerto contra huésped (EICH): en este caso, el sistema inmunitario donado reacciona contra las células del propio organismo. Esto puede ocurrir de forma breve (aguda) o más tarde y de forma prolongada (crónica), por lo que, en determinadas circunstancias, puede ser necesario suprimir el sistema inmunitario de forma permanente (terapia inmunosupresora).
5.4 Otras opciones terapéuticas
Danazol
Algunos pacientes con anemia aplásica padecen un trastorno congénito poco frecuente en el que los extremos de los cromosomas (telómeros) se acortan, lo que se denomina telomeropatía. El acortamiento de los telómeros provoca un trastorno en la división celular y, por lo tanto, una disminución de la formación de células sanguíneas en la médula ósea.
Se ha demostrado que el danazol, una variante sintética de la hormona sexual masculina testosterona, puede provocar un alargamiento de los telómeros, lo que puede conducir a una mejora de los síntomas e incluso a la normalización de la formación de sangre.
Eltrombopag (Revolade®)
Desde 2015, el eltrombopag está autorizado para pacientes adultos con anemia aplásica adquirida (AAA) grave si
- no han respondido a un tratamiento inmunosupresor previo o
- han recibido un tratamiento previo intenso y
- no son aptos para un trasplante de médula ósea o de células madre.
Debido a las excelentes tasas de respuesta en los ensayos clínicos, el eltrombopag ya ha sido autorizado en EE. UU. en combinación con hATG y CsA para el tratamiento de primera línea de la anemia aplásica. No existe autorización para el tratamiento de primera línea en la UE. Sin embargo, debido a su eficacia demostrada, la Sociedad Alemana de Hematología y Oncología Médica recomienda el uso de eltrombopag en el tratamiento de primera línea de la SAA/vSAA adquirida en el marco de sus directrices.
El eltrombopag actúa sobre la regulación de la formación de células madre sanguíneas y plaquetas. El medicamento activa la trombopoyetina, que controla la formación de plaquetas y la hematopoyesis (hematopoyesis). La dosis eficaz para la AAG es de
150 mg/día. Se ha demostrado que el uso de eltrombopag mejora los valores de plaquetas, eritrocitos y neutrófilos en la mayoría de los pacientes. Asimismo, el eltrombopag provocó una mejora o normalización de la celularidad de la médula ósea. En pacientes que anteriormente necesitaban transfusiones regulares, se prolongó el número de días hasta la siguiente transfusión o dejaron de necesitar transfusiones.
Otros
Deben evitarse los tratamientos sin eficacia demostrada, como la monoterapia con esteroides o la monoterapia con factores de crecimiento hematopoyéticos, ya que solo suponen una pérdida de tiempo y pueden empeorar considerablemente la situación inicial del paciente con respecto a las opciones terapéuticas probadas.
5.5 Terapia de apoyo (de apoyo)
La prevención y el tratamiento de infecciones, la prevención de hemorragias, la estrategia de transfusión adaptada individualmente y el tratamiento de la sobrecarga de hierro son de especial importancia en la terapia de apoyo. Estas medidas han mejorado en los últimos años y contribuyen a aumentar la probabilidad de supervivencia, incluso si el tratamiento no logra una respuesta completa.
Infecciones
En caso de infecciones febriles, se debe acudir al médico lo antes posible para que establezca un diagnóstico e inicie el tratamiento.
En determinados casos, puede ser conveniente el uso preventivo de antibióticos contra las bacterias y de antimicóticos contra los hongos.
- En pacientes con neutropenia grave (recuento de granulocitos/neutrófilos < 0,5 G/l)
- En pacientes en tratamiento con antitimocito globulina (ATG) o alemtuzumab, se debe llevar a cabo una prevención adicional contra otros patógenos (por ejemplo, Pneumocystis jirovecii, citomegalovirus).
Además, en caso de recuento bajo de granulocitos/neutrófilos (< 0,5 G/l), deben observarse diversas medidas de comportamiento:
- Evitar el contacto con personas que padezcan infecciones.
- Evitar el contacto físico cercano con animales.
- Evitar las grandes aglomeraciones de personas, especialmente en los meses de invierno.
- Tomar las medidas de higiene habituales, por ejemplo, lavarse las manos, cuidar la higiene bucal, prestar atención a la frescura y la limpieza de los alimentos crudos.
- Evitar el contacto cercano con esporas de hongos, especialmente en la jardinería, no limpiar el contenedor de residuos orgánicos ni remover el compost.
En casos muy raros, por ejemplo, en infecciones graves, se puede considerar el uso de factores de crecimiento hematopoyéticos G-CSF o GM-CSF para estimular las defensas inmunitarias del organismo o la transfusión de concentrados de glóbulos blancos (concentrados de granulocitos).
Encontrará más información en el folleto «¿Infecciones? ¡No, gracias!» de M. Exner y A. Simon, de la Asociación Alemana de Ayuda contra la Leucemia y el Linfoma.
Hemorragias
Debido a la reducción del número de plaquetas, los pacientes con anemia aplásica pueden sufrir hemorragias graves, incluso mortales. Puede producirse un aumento o una prolongación de la hemorragia en caso de lesiones o operaciones, así como hemorragias repentinas sin causa aparente.
Por eso es tan importante no limitar la función de las plaquetas existentes con medicamentos. Entre estos medicamentos se encuentran, por ejemplo, los inhibidores de la agregación plaquetaria, ya que impiden la aglutinación de las plaquetas y, por lo tanto, dificultan la coagulación de la sangre.
Por eso es tan importante no limitar la función de las plaquetas existentes con medicamentos. Entre estos medicamentos se encuentran, por ejemplo, los inhibidores de la agregación plaquetaria, ya que impiden la aglutinación de las plaquetas y, por lo tanto, interfieren en la coagulación sanguínea. Por lo tanto, el uso de antiagregantes plaquetarios como el ácido acetilsalicílico (AAS) debe considerarse extremadamente crítico, especialmente en casos de recuento plaquetario muy bajo, y debe sopesarse cuidadosamente.
En mujeres con menstruaciones muy abundantes, para evitar una pérdida excesiva de sangre en caso de trombocitopenia, se puede suspender temporalmente la menstruación mediante terapia hormonal, por ejemplo, con la administración continua de la píldora o una inyección trimestral.
El medio esencial para evitar hemorragias y tratar las hemorragias existentes es la transfusión de concentrados de plaquetas.
Transfusiones de sangre
Las transfusiones son necesarias en muchos pacientes para garantizar una resistencia física y una calidad de vida suficientes, así como para evitar complicaciones hemorrágicas. En caso de síntomas correspondientes (anemia, hemorragias), pueden sustituir temporalmente las células sanguíneas que faltan. No se transfiere (transfunde) toda la sangre, sino solo el tipo de célula que se necesita (glóbulos rojos o plaquetas).
Para producir un concentrado, tras una donación de sangre, primero se analiza la sangre para descartar infecciones transmisibles, luego se eliminan los glóbulos blancos y, a continuación, se separan y concentran los diferentes componentes sanguíneos. Los familiares no pueden ser donantes de sangre, ya que estas donaciones específicas conllevan riesgos especiales.
Transfusión de glóbulos rojos/concentrados de eritrocitos (transfusión de EK)
Para garantizar una buena compatibilidad, el preparado utilizado no solo se selecciona en función del grupo sanguíneo (A, B, AB, 0) y del factor Rh, sino que cada preparado se prueba individualmente para cada paciente. Para ello, en una prueba de compatibilidad, denominada prueba cruzada, se mezcla la sangre del paciente con la sangre del concentrado de eritrocitos (EK) y se examina. Especialmente si hay anticuerpos, puede llevar más tiempo encontrar un concentrado de eritrocitos adecuado. Si un paciente es negativo al virus de la citomegalía (CMV) y existe la posibilidad de un trasplante posterior de médula ósea o células madre, se deben administrar concentrados negativos al CMV.
En general, los concentrados de eritrocitos deben utilizarse con precaución, ya que pueden provocar una sobrecarga de hierro en el organismo. Las indicaciones para una transfusión son
- una disminución pronunciada del rendimiento asociada a fatiga o en el contexto de dificultad respiratoria, por ejemplo, durante el esfuerzo físico, y en función de las comorbilidades respectivas, por ejemplo, insuficiencia cardíaca
- un nivel muy bajo de hemoglobina (< 8 g/dl)
En general, en lo que respecta a la terapia transfusional se aplica lo siguiente:
¡Tanto como sea necesario, tan poco como sea posible!
Por lo tanto, la indicación de la transfusión debe evaluarse individualmente para cada paciente en función de los siguientes puntos:
- Síntomas clínicos (por ejemplo, frecuencia cardíaca, frecuencia respiratoria, dificultad respiratoria, signos de hemorragia, fiebre, infección…)
- molestias subjetivas (por ejemplo, disminución significativa del rendimiento, cansancio, debilidad, dolor de cabeza, palpitaciones en
los oídos…) - enfermedades concomitantes/antecedentes individuales (por ejemplo, insuficiencia cardíaca, hemorragia)
- Medicamentos/terapias (por ejemplo, terapia con ATG, intervención quirúrgica)
- Posibilidad de monitorización y transfusión a corto plazo (ambulatoria/hospitalaria)
La estrategia de transfusión restrictiva se aplica principalmente a los pacientes en los que se ha planificado un trasplante alogénico de células madre. En caso de un trasplante de células madre planificado, no se deben realizar transfusiones de productos sanguíneos de familiares.
Transfusión de concentrados de plaquetas/trombocitos (transfusión de TK)
En caso de hemorragia, se pueden transfundir concentrados de plaquetas (concentrados de trombocitos, TK) para evitar complicaciones. Dado que las hemorragias con recuentos bajos de trombocitos pueden ser emergencias agudas que ponen en peligro la vida, en estos casos se debe actuar de inmediato. Si los recuentos de trombocitos son muy bajos, también se pueden administrar concentrados de trombocitos de forma preventiva. La vida útil de las plaquetas es de solo unos pocos días. Por lo tanto, si la producción de trombocitos en la médula ósea es muy baja o inexistente, puede ser necesario administrar concentrados de trombocitos varias veces por semana.
Los trombocitos llevan marcadores HLA, que son diferentes para cada persona. En algunos pacientes se producen anticuerpos contra estos marcadores HLA. Esto puede ocurrir de forma espontánea, en el contexto de enfermedades o después del embarazo. Si existen estos anticuerpos HLA, las plaquetas transfundidas se destruyen inmediatamente y no se produce un aumento suficiente de plaquetas después de la transfusión de un concentrado de plaquetas. Para estos pacientes, es necesario producir concentrados de plaquetas especiales compatibles con HLA a partir de donantes con características HLA adecuadas.
La indicación para una transfusión de concentrados de plaquetas existe:
- cuando los valores plaquetarios son < 20 G/l y hay fiebre > 38 °C, infecciones, signos de hemorragia o antecedentes de hemorragias graves (grado 3 o 4 de la OMS), así como en caso de aloinmunización (anticuerpos HLA, véase más arriba)
- si no existen riesgos que aumenten el peligro de hemorragia (por ejemplo, fiebre, infecciones, antecedentes de hemorragias graves, aloinmunización), también se puede realizar la transfusión cuando los valores plaquetarios sean < 5 G/l. Los requisitos para ello son controles regulares y frecuentes (por ejemplo, al menos una vez a la semana), la ausencia de signos de hemorragia (por ejemplo, petequias) y la posibilidad de realizar una transfusión rápida en caso de signos de hemorragia.
«Límite individual del paciente»: muchos pacientes tienen un límite estable por debajo del cual se producen signos de hemorragia más graves. - Antes y durante el tratamiento con ATG, el recuento plaquetario debe elevarse a 50 G/l, ya que la infusión de ATG puede provocar una rápida disminución de las plaquetas.
- Antes de cualquier operación o intervención, es imprescindible informar al médico responsable del tratamiento sobre la anemia aplásica y, a ser posible, proporcionarle un hemograma actualizado.
Sobrecarga de hierro
Con cada concentrado de eritrocitos se absorbe más de 100 veces la cantidad de hierro que se ingiere diariamente con los alimentos. Dado que el cuerpo humano no puede eliminar activamente el hierro, este se almacena en diversos órganos, especialmente en el hígado, el corazón, los riñones y la médula ósea (véase la tabla 2), y puede dañarlos. Los pacientes con anemia aplásica o síndrome mielodisplásico (SMD) corren el riesgo de sufrir una sobrecarga de hierro incluso sin transfusiones, ya que la mala función de la médula ósea reduce la formación de nuevos eritrocitos y el hierro no puede utilizarse completamente para la formación de nuevos glóbulos rojos.
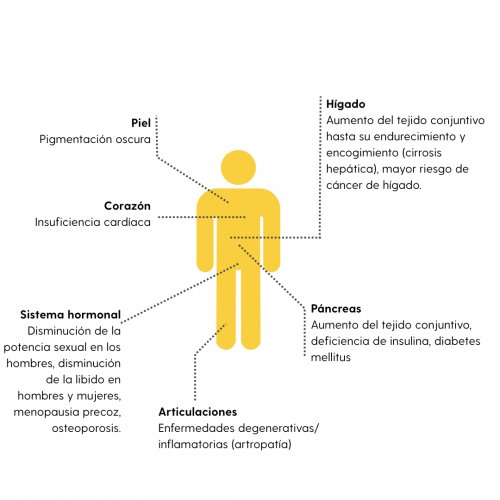
Complicaciones de la sobrecarga de hierro
Por lo general, en los primeros meses tras el diagnóstico aún no se alcanzan valores de hierro almacenado (ferritina) o hierro hepático que requieran un tratamiento inmediato para la unión y eliminación del exceso de hierro (terapia con quelantes). Por lo tanto, se debe esperar al menos 6 meses tras el inicio de la inmunosupresión. En caso de necesidad continua de transfusiones regulares, se recomienda la terapia de quelación cuando los niveles de ferritina sérica superan los 1000 µg/l. Esto se aplica especialmente a los candidatos a trasplante, ya que la sobrecarga de hierro se asocia con una mayor mortalidad relacionada con el trasplante y una mayor incidencia de enfermedades (morbilidad).
Los medicamentos que se utilizan actualmente para tratar la sobrecarga de hierro son, en general, bien tolerados. Entre los efectos secundarios más importantes se encuentran las náuseas, la diarrea y la disfunción renal, que, sin embargo, desaparecen tras suspender el tratamiento.
Si la ferritina sérica es inferior a 1000 µg/l, en caso de sobrecarga de hierro relacionada con transfusiones, se puede considerar la interrupción del tratamiento en función de las necesidades transfusionales individuales. No obstante, esto siempre debe hacerse tras consultar con el médico responsable del tratamiento.
Si no se necesitan transfusiones y el nivel de hemoglobina es suficientemente alto, se recomienda la terapia de sangrado (cantidad individual, por ejemplo, con cantidades pequeñas como 100 ml por extracción) como una opción terapéutica eficaz y con pocos efectos secundarios para la sobrecarga de hierro.
En general, la sobrecarga de hierro debe controlarse periódicamente y ajustarse el tratamiento.
Encontrará más información en el folleto «Sobrecarga de hierro relacionada con transfusiones» de la Asociación Alemana de Ayuda contra la Leucemia y el Linfoma.
Actividades
En pacientes con anemia aplásica, la actividad física y el ejercicio son recomendables en función de los valores sanguíneos y el estado de salud. Sin embargo, se debe tener cuidado de no sobreesfuerzarse. Por lo tanto, es aconsejable practicar deporte bajo control del pulso. Esto es especialmente importante en caso de anemia, ya que, cuando hay una disminución de los eritrocitos, el cuerpo suele intentar compensar esta deficiencia aumentando el ritmo cardíaco, lo que puede suponer una carga excesiva para el corazón. En caso de trombocitopenia, se deben evitar sin falta los deportes que conllevan riesgo de lesiones, como las artes marciales o la escalada.
Rehabilitación
Si, debido a la anemia aplásica, ya no es posible llevar una vida «normal» como de costumbre, puede ser conveniente realizar medidas de rehabilitación, fisioterapia ambulatoria o terapia física, o recibir atención psicológica o psicoterapéutica. Estas medidas deben adaptarse individualmente a cada paciente.
Si se han planificado medidas terapéuticas intensivas, es recomendable realizar las medidas de rehabilitación después de estas terapias. La gimnasia terapéutica o la fisioterapia, así como la asistencia psicológica o psicoterapéutica, también son útiles como complemento de la terapia.
En la reintegración de un niño con anemia aplásica tras completar la terapia inmunosupresora o el trasplante, puede ser conveniente una medida orientada a la familia en un centro de seguimiento oncológico pediátrico, debido a la elevada carga psicosocial que supone para las familias.
6 Pronóstico
Cuanto mayor sea el recuento de granulocitos y menor la edad del paciente en el momento del diagnóstico, mejor será el pronóstico.
Los datos sobre la supervivencia tras diferentes terapias que se mencionan a continuación son datos estadísticos. Esto significa que no se pueden aplicar automáticamente a cada paciente. Esta lista solo pretende ofrecer una visión general de cómo han mejorado las posibilidades y la supervivencia en los últimos años. A menudo hay subgrupos que no se tienen en cuenta aquí. En todas las formas específicas de terapia, los resultados para los pacientes menores de 20 años son significativamente mejores que para los pacientes mayores de 20 años. Lo mismo se aplica a los pacientes menores de 40/50 años en comparación con los pacientes mayores de 40/50 años. En los trasplantes de células madre, los resultados son considerablemente mejores cuando el donante proporciona células madre directamente de la médula ósea y no las células madre periféricas obtenidas de la sangre.
Según los datos publicados, la supervivencia global en la SAA/vSAA después de 3-6 años y separada por las diferentes formas de terapia específicas es la siguiente:
- después de un TCS alogénico de un donante familiar HLA compatible: 75-90 %
- tras un TCS alogénico de donantes no emparentados HLA idénticos: 65-73 %
- tras un tratamiento con ATG/CsA: 76-96 %
7 Registro
Los pacientes con evidencia de un clon de HPN pueden ser incluidos en el Registro Internacional de HPN (PNH Registry) a través de la Clínica Universitaria de Essen, con el fin de obtener más información sobre este grupo de pacientes con AA.
Si está interesado o tiene alguna pregunta, póngase en contacto con el Prof. Dr. med. Alexander Röth (alexander.roeth(at)uk-essen.de) por correo electrónico.
Los pacientes con telomeropatía pueden ser incluidos en el registro AA-BMF de la Clínica Universitaria de Aquisgrán, con el fin de obtener información sobre este subgrupo de pacientes con AA.
Si está interesado o tiene alguna pregunta, póngase en contacto con el Prof. Dr. med. Tim H. Brümmendorf (tbruemmendorf(at)ukaachen.de) por correo electrónico.