
APLASTIC ANAEMIA (AA)
1 What is AA?
- The number of cells in the bone marrow (cellularity) is less than 25% of that in healthy bone marrow, based on a bone marrow biopsy. Cell production may be reduced during the course of the disease (hypoplastic) or completely absent (aplastic).
- Reduction of two (bicytopenia) or three cell lines (tri- or pancytopenia) of varying degrees in the blood film.
- There is no evidence of (new) development of connective tissue in the bone marrow (fibrosis) or invasion of the bone marrow by malignant cells or cells outside the bone marrow.
- No radiotherapy or chemotherapy has been carried out recently that might explain a disorder of bone marrow function (bone marrow insufficiency).
- In addition, there have been no significant cell changes (dysplasia) in blood development (haematopoiesis).
- non-severe aplastic anaemia = nSAA (“non-severe AA”)
- severe aplastic anaemia = SAA (“severe AA”)
- very severe aplastic anaemia = vSAA (“very severe AA”)
1.1 General
Aplastic anaemia is a non-malignant haematological disease. It is due to a disorder of bone marrow function that results in reduced development of blood cells.
Aplastic anaemia can be divided into congenital forms (e.g. Diamond-Blackfan or Fanconi anaemia) and acquired forms, depending on the age at which it occurs. Acquired forms can occur at any age.
1.2 Occurrence (epidemiology)
The incidence of aplastic anaemia in Central Europe is 2-3 new cases per million people per year. Aplastic anaemia is therefore a very rare disease. Most people who are affected fall ill between the ages of 10 and 25 or above the age of 60, with both sexes being equally affected.
1.3 Origin (pathogenesis)
Studies have shown that a subtype of lymphocyte in the body’s own immune system attacks cells in the bone marrow, and this autoimmune process prevents the development of new blood cells.
In most cases, it is not possible to identify the cause of aplastic anaemia, so the origin of the disease is still unknown (idiopathic). In some cases, medications, toxic substances or viral infections are thought to be the cause.
1.4 Diagnostic criteria and classification
In order to classify a disease as aplastic anaemia, the following criteria must be met:
Aplastic anaemia is subdivided according to blood values (see table below) into:
and is of crucial importance for prognosis and treatment.
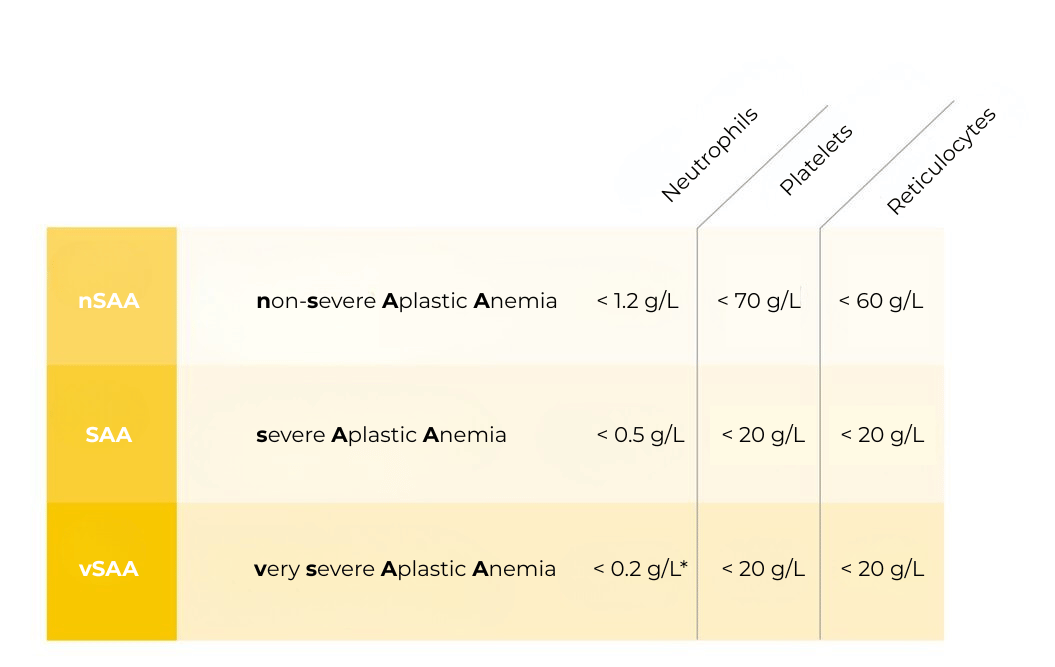
Classification of aplastic anaemia based on the blood count (cell count and film) Two out of three criteria must be met *For classification as vSAA, the criterion granulocytes < 0,2 G/l must be met.
2.1 Anaemia
A reduction in oxygen-transporting red blood cells (erythrocytes) can cause weakness, fatigue and shortness of breath and even palpitations, especially during physical exertion. In addition, patients with anaemia often show paleness, especially in the palms of the hands, although the presence of paleness is not evidence of anaemia.
2.2 Increased susceptibility to infection
A reduced number of white blood cells (leukocytes) increases the risk of infection. Since the body’s own defence system does not function sufficiently with a reduced number of neutrophils, a subtype of white blood cells, such an infection can take a life-threatening course within hours and lead to blood poisoning (septicaemia).
It is therefore important that you inform your doctor immediately if you develop a fever. A fever is defined as a body temperature of over 38°C measured in the ear twice within an hour or over 38.3°C measured once in the ear.
2.3 Bleeding
If the number of blood platelets (thrombocytes) is reduced, blood clotting may be impaired. This can lead to bleeding gums and petechiae, small punctiform bleedings in the skin, or bruises (haematomas). These can also occur spontaneously, i.e. without previous injury. In the case of impaired clotting, even a relatively slight bleeding or injury (e.g. during a visit to the dentist) can be serious. In the event of bleeding, you should therefore contact your doctor as soon as possible so that he can decide whether special measures (e.g. platelet transfusion) are necessary.
- Medical history (anamnesis), including the family history and a detailed record of any medications taken
- Physical examination, e.g. signs of anaemia or bleeding
- Cell studies
-
- Microscopic differential blood count
- Reticulocytes
- PNH diagnostics (a PNH clone is detectable in up to 70% of AA cases), see Chapter 4.3 PNH, 3 Diagnosis
-
- Clinical chemistry
-
- Haemolysis parameters: in particular LDH, haptoglobin, bilirubin
- Coagulation: quick value, PTT, fibrinogen
- Liver function parameters: AST, ALT and AP
- Renal function parameters: creatinine, uric acid
- Blood sugar
- Total protein, electrophoresis, immunoglobulins
- Inflammation parameters CRP
- Vitamin B12 and folic acid levels
- Iron status: ferritin. At ferritin values > 1000 ng/ml further clarification of possible organ damage due to possible iron overload
- Virus diagnostics: hepatitis A, B, C; HIV, EBV, CMV, Parvovirus B19
- Antinuclear and anti-DNA antibodies
-
- Functional diagnostics
-
- Ultrasound (sonography) of heart and upper abdomen
- X-ray examination of the chest (thorax)
- ECG
-
- Special investigations
-
-
- HLA typing of the patient and his siblings
- In cases of suspected “congenital” bone marrow insufficiency syndrome, further diagnostic tests, e.g. chromosome breakage analysis in case of suspected Fanconi anaemia, telomere length determination if telomeropathy is suspected, genetic tests
-
- Exclude other diseases
- Discover the possible causes (aetiology)
- Determine the severity of the aplastic anaemia
- Determine the prognosis
If one or more of the above complaints and symptoms are present, the family doctor will have a blood test done. If this reveals an irregularity in the blood count, the patient may be referred to a specialist in haematology and/or oncology..
A number of further examinations will be carried out in this case:
If a reduced number of one or more blood cell lines is confirmed without any known cause for increased damage or breakdown of these blood cells, an urgent bone marrow examination should be carried out. This will help to determine whether there is a disorder of blood development or another causeegt.
For this purpose, a bone marrow puncture is performed, which can be done on an outpatient basis. A bone cylinder is usually taken from the pelvis under local anaesthetic using a hollow needle (Jamshidi needle) (bone marrow biopsy, bone marrow punch). This cylinder is approx. 1.5 cm in length with a diameter of 2-3 mm and is examined and evaluated under a microscope (histology).
In addition, blood, bone marrow and yellow marrow fragments are obtained with bone marrow aspiration. These are spread out over a glass microscope slide, dried and stained. They are then assessed under the microscope, whole and in their position in relation to each other (cytological examination).
Furthermore, genetic tests can be carried out on the bone marrow cells, and the results of these tests can help to distinguish between diagnoses.
As the individual laboratory steps in preparing the bone marrow histology are time-consuming, it takes about 1-2 weeks to obtain a complete result. If the development of two or three cell lines (erythrocytes, leukocytes, platelets) is impaired according to the diagnostic criteria in Section 1.4 without the presence of pathologically altered cells (e.g. leukaemia cells) and without prior chemotherapy or radiotherapy, the disease is referred to as aplastic anaemia
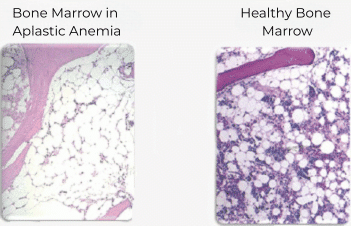
Bone marrow in a patient with aplastic anaemia compared with healthy bone marrow. In the diseased bone marrow one can see mostly connective tissue and fat cells. In the healthy marrow, the blood cells stand out from the large white fat cells as small coloured dots.
The aim of these numerous investigations is to:
In patients with severe or very severe aplastic anaemia who are in good physical condition, it is advisable to carry out HLA typing of the patient as soon as the condition has been diagnosed. If the patient has siblings, they can also be typed to determine their suitability for stem cell donation.
Without specific therapy, aplastic anaemia is fatal in up to 70% of cases in adulthood.
Aplastic anaemia may develop into myelodysplastic syndrome (MDS) or acute myeloid leukaemia (AML). In addition, some AA patients have a PNH-specific mutation.
- Very severe (vSAA) and severe aplastic anaemia (SAA)
- Non-severe aplastic anaemia (nSAA) with a marked reduction in at least one cell line (cytopenia), which requires regular transfusions or leads to a risk of infection or bleeding
- Transition (progression) from an nSAA to an SAA
- Patients with vSAA or SAA > 40-50 years
- Patients without HLA-identical (sibling-) donors
- Patients with nSAA at risk of severe cytopenia in at least one cell row
- Toxic side effects during conditioning therapy
- Infections
- Graft versus Host-Disease (GvHD)
- Transplant rejection
- have not responded to previous immunosuppressive therapy, or
- have received intensive pre-treatment and
- are not suitable for bone marrow or stem cell transplantation.
- Pronounced reduction in stamina associated with fatigue or in the context of shortness of breath, e.g. under physical exertion, and depending on the respective concomitant diseases, e.g. heart failure
- A very low haemoglobin level (< 7 g/dl)
- Avoid contact with people who have infections
- Avoid close physical contact with animals
- Avoid large crowds, especially in the winter months
- Carry out usual hygiene measures, e.g. washing hands, oral hygiene, and ensure freshness and cleaning of raw food
- Avoid close contact with fungal spores, especially gardening, do not clean organic waste bins or turn compost
5.1 Overview
Haematological spontaneous healing (spontaneous remission) only occurs very rarely in cases of severe bone marrow failure.
Treatment is needed for:
If the patient has nSAA, one can usually apply ‘watchful waiting’ without intensive therapy.
While a few decades ago there was hardly any prospect of a cure or long-term improvement, there are now promising options. Two main types of treatment are available: immunosuppressive therapy (IST), and stem cell transplantation (SCT) or bone marrow transplantation (BMT). In addition, there are specific treatments for certain subgroups of patients. Which of these is appropriate depends on the severity of the disease, the patient’s age and possible concomitant diseases — as well as the degree of the HLA match (HLA compatibility) with the potential bone marrow donor, whether related or unrelated.
If therapy is indicated, treatment should be started as soon as possible to avoid the progression of the disease and its possible complications (e.g. pronounced anaemia, infections, bleeding and coagulation disorders). Early therapy planning in cooperation with a specialised centre is therefore important.
The course of therapy for patients with severe or very severe aplastic anaemia and for patients with nSAA who require treatment is shown in the figure below:
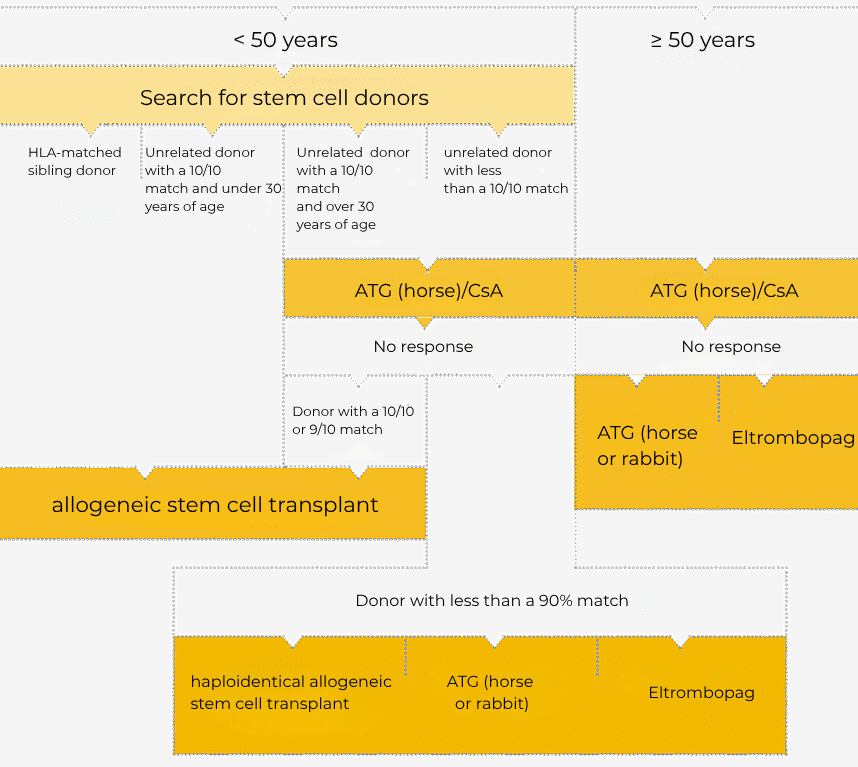
Clearly simplified presentation of the therapy algorithm. For the complete algorithm, please visit Onkopedia (German)
5.2 Immunosuppressive Therapy (IST)
Antithymocyte Globulin (ATG) and ciclosporin (CsA)
Since in aplastic anaemia the body’s own immune system turns against its own bone marrow, immunosuppressive therapy is often indicated, especially in the case of:
Immunosuppressive therapy is usually a combination of the drugs antithymocyte globulin and ciclosporin. This allows the bone marrow to recover. In the course of the therapy, the blood count usually deteriorates for a short time before there is an improvement. The most common complications are fever or allergic reactions and rarely infections.
ATG is an antibody that destroys the overactive, bone marrow-damaging T lymphocytes. ATG is usually given for 4-5 days as an infusion into a large vein via a central venous catheter (CVC). During ATG therapy, the platelet count should be elevated to 30 G/l or kept there by means of platelet transfusion, if necessary, as the platelet levels can drop rapidly during therapy. For an ATG therapy, an inpatient stay of about 1-2 weeks must be expected. Side effects of ATG therapy can be allergic reactions such as skin rash and fever. To suppress acute side effects of ATG, a cortisone preparation, e.g. prednisone or prednisolone, is also administered for a short time.
Current data show that serum obtained from horses (equine ATG, horse ATG, hATG) is significantly more effective than rabbit ATG (rATG). However, the only approved horse ATG (hATG) preparation (Lymphoglobulin®) was withdrawn from the market in the EU in 2007, so the medication must currently be imported from outside the EU. Due to the lack of approval in the EU, it is advisable to clarify the responsibility for costs in advance with the respective health insurance company. A new approval of equine/horse ATG in the EU has been applied for by the manufacturer, but not yet decided.
Ciclosporin, which inhibits the release of immunostimulants, is another key factor in the therapeutic response to the disease. CsA is subject to regular laboratory tests to ensure that the optimal effect is achieved by adjusting the dose, if necessary. The aim is to achieve a minimum concentration of 100-200 ng/ml in the blood. For a stable effective level, the medication should be taken very regularly at fixed intervals of 12 hours.
Possible side effects of CsA therapy are infections, deterioration in kidney function, increased blood pressure, gum growth (gingival hyperplasia), increase in hair growth or tremor.
CsA is taken as a capsule or juice for at least 12 months. To ensure a good and stable response to therapy, it is important that discontinuation involves reducing the dose of CsA very slowly and gradually in order to prevent a relapse. However, in some patients, CsA needs to be given for a longer period or even permanently to maintain the success of the therapy.
By intensifying immunosuppressive therapy, a cure (complete remission, CR) or at least a marked improvement (partial remission, PR) can be achieved in about 50-75% of patients, with transfusion independence and a significant reduction in the risk of infection and bleeding. It takes about 2-4 months, in some patients even 6 months, until an improvement of the blood values occurs. In most cases complete normalisation of blood values cannot be achieved.
If there is no response, a repeat of the immunosuppressive therapy can be considered after 4-6 months.
The risk of a relapse of the disease (recurrence) was about 35% earlier, when slow CsA tapering was still uncommon. The risk of relapse is lower with very slow CsA reduction. In the event of a relapse, a repeat of the immunosuppressive therapy is possible, as the chance of a renewed response is 30-60%.
In addition to specific therapy, every patient should receive supportive therapy, see usupportive therapy .
Alemtuzumab
There are also other drugs that work through the same mechanism of immunosuppression. These include alemtuzumab, an antibody that works against T lymphocytes. This drug is used to treat chronic lymphocytic leukaemia (CLL) or multiple sclerosis (MS), but it has also shown good response rates in trials of aplastic anaemia, especially in older patients. One advantage of this drug is that it is only injected under the skin, so no hospital stay is necessary. If the patient previously had an infection with the cytomegalovirus (CMV), this blood value should be checked regularly, as this viral infection can reoccur under therapy.
Patients who did not respond to other therapies showed response rates of 37-48% when treated with alemtuzumab.
5.3 Allogeneic transplantation
In patients up to the age of approximately 50 years with severe or very severe aplastic anaemia (SAA or vSAA) and availability of a sibling donor who is fully compatible with the patient with respect to compatibility markers (HLA) (HLA-identical), the preferred treatment (first-line therapy) is an allogeneic transplantation.
Patients younger than 18 years of age can also receive stem cells from an unrelated HLA-identical donor (foreign donor) if they do not have an HLA-identical family donor. It is important that ‘fine mapping’ is performed and that donor and recipient are completely identical in this regard.
In recent years, the complication rate for HLA-identical foreign donor transplantation has been significantly reduced, so that it is increasingly used – especially in patients up to 40 years of age who do not respond to immunosuppressive treatment.
The aim of allogeneic transplantation is to replace the patient’s non-functional bone marrow with healthy stem cells from a donor. To achieve this, the patient’s bone marrow is first destroyed by various measures (chemotherapy, antibody therapy, radiation). This ‘conditioning’ is carried out in the days immediately before the transplantation.
In parallel, new, healthy stem cells are collected from a healthy, related or unrelated volunteer.
Stem cells can be obtained directly from the bone marrow with bone marrow biopsy under anaesthesia. The punctures in the iliac crest for bone marrow collection may cause bruising and pain that may last for several days. In addition, there is the general anaesthetic risk.
Alternatively, the donor is injected with a drug that stimulates granulocyte development (G-CSF) over several days. The increased number of blood stem cells produced in this way migrate from the bone marrow into the blood. These ‘peripheral blood stem cells’ (PBSC) are then removed with a special device (apheresis), as in a blood plasma donation. The procedure can lead to flu-like symptoms and pain.
If the stem cells are obtained directly from the bone marrow, this is called a bone marrow transplantation (BMT). If the stem cells are obtained by apheresis, this is called a stem cell transplantation (SCT).
Studies suggest that treatment of aplastic anaemia with stem cells from peripheral blood may be associated with increased complications such as acute or chronic rejection. If possible, stem cells obtained directly from the bone marrow should therefore be used.
Regardless of the method of obtaining the stem cells, they are purified and examined for infectious agents. The patient then receives the healthy stem cells. The transplantation itself is like a blood transfusion. If everything goes well, the donor stem cells “grow” and lead to normal bone marrow function and blood development. An inpatient stay of at least four weeks is required for a transplant.
During the transplantation, the patient receives prophylactic medication to prevent infections caused by bacteria and fungi. In addition, a cortisone preparation, e.g. prednisolone, and ciclosporin (CsA) are administered to influence the immune system for several months.
Potential complications due to the transplantation are:
Graft versus Host-Disease (GvHD): Here the donated immune system reacts against the body’s own cells. This can be short term (acute) or long lasting (chronic), so that under certain circumstances permanent suppression of the immune system (immunosuppressive therapy) may be necessary.
5.4 Further treatment options
Danazol
Some patients with aplastic anaemia have a rare congenital disorder in which the ends of the chromosomes (telomeres) are shortened, which is called telomeropathy. The shortening of the telomeres results in disrupted cell division and thus to reduced development of blood cells in the bone marrow.
Danazol, a synthetic variant of the male sex hormone testosterone, may cause the telomeres to lengthen, which can lead to an improvement in symptoms and even normal blood development.
Eltrombopag (Revolade®)
Eltrombopag has been approved for adult patients with acquired severe aplastic anaemia (SAA) since 2015 if they:
Eltrombopag acts to control the development of blood stem cells and platelets. The medication activates thrombopoietin, which controls the development of platelets and blood cells (haematopoiesis). The initial dose is 50 mg/day (maximum dose 150 mg/day). In the authorisation trial, it was shown that the use of eltrombopag led to an improvement of platelet, erythrocyte and neutrophil levels in some patients. Eltrombopag resulted in an improvement or normalisation of bone marrow cellularity. Patients who had previously required regular transfusions had an increased number of days until the next transfusion or became transfusion-independent. Due to the very good response rates in clinical trials, eltrombopag has already been approved in the USA in combination with hATG and CsA for first-line treatment of aplastic anaemia. An application for approval in the EU has also been filed.
Other
Therapies without proven efficacy, e.g. steroid monotherapy or monotherapy with haematopoietic growth factors, should be avoided, as they only mean loss of time and can significantly worsen the initial situation of the patient with regard to one of the proven therapeutic options.
5.5 Supportive Therapy
Blood Transfusions
Transfusions are necessary for many patients to ensure adequate physical fitness and quality of life and to avoid bleeding complications. They can temporarily replace the missing blood cells in case of corresponding symptoms (anaemia, bleeding). Not all of the blood is transfused, but only the type of cell that is needed (red blood cells or platelets).
To do this, the blood is examined after a blood donation to rule out transmissible infections. The white blood cells are then removed, and finally the various blood components are separated and concentrated. Family members are not allowed to donate blood, as these so-called directed donations pose particular risks.
In order to ensure satisfactory tolerance, the preparation used is not only selected according to blood group (A, B, AB, O) and rhesus factor, but each individual preparation is tested individually for each patient. For this purpose, in a compatibility test or ‘cross-matching’, the patient’s blood is mixed with blood from the packed red blood cells (PRBCs) and examined. Especially if antibodies are present, it can take longer to find suitable PRBCs. If a patient is cytomegalovirus (CMV) negative and there is a possibility of a later bone marrow or stem cell transplantation, CMV-negative concentrates should be given.
In general, packed red blood cells should be used with caution, as they can lead to the body being overloaded with iron. Indications for a transfusion are:
Each unit of packed red blood cells absorbs more than 100 times the amount of iron than is taken in daily with food. As the human body cannot actively excrete iron, it is deposited in various organs, especially the liver, heart, kidney and bone marrow (see Table 2) and can damage them. Patients with aplastic anaemia or myelodysplastic syndrome (MDS) are particularly at risk because the impaired bone marrow function already decreases the development of new red blood cells and the iron cannot be used completely for the development of new ones.
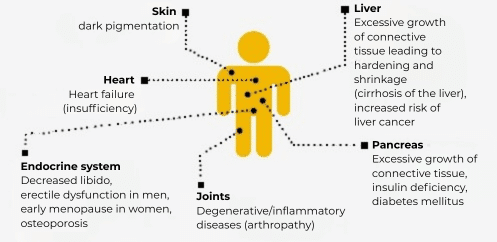
Complications of iron overload
As a rule, in the first few months after diagnosis, no storage iron (ferritin) or liver iron levels are reached which require immediate therapy to bind and excrete the excess iron (chelation therapy). It is therefore advisable to wait at least 6 months after initiating immunosuppression. If there is a continuing need for regular transfusions and serum ferritin levels are above 1,000 µg/l, chelation therapy is indicated. This is especially true for transplant candidates, as iron overload is associated with higher transplant-related mortality and morbidity.
The drugs used today to treat iron overload are generally well tolerated. The major side effects include nausea, diarrhoea and renal dysfunction, but these disappear after discontinuation.
If the serum ferritin is permanently below 500 µg/l and there is transfusion-related iron overload, interrupting treatment may be considered depending on individual transfusion needs. However, this should always be done in consultation with the treating doctor.
Further information on this issue can be found in the Brochure „Transfusion-Induced Iron Overload” (German) by the Deutsche Leukämie- & Lymphom-Hilfe.
Infections
In the case of febrile infections, a doctor should be consulted as soon as possible to make a diagnosis and initiate therapy.
In certain cases, the preventive use of antibiotics aghttps://www.leukaemie-hilfe.de/fileadmin/user_upload/DLH_Broschuere_Eisenueberladung_2024_lowres.pdf ainst bacteria and of antimycotics (‘against fungi’) can be useful.In addition, if the granulocyte/neutrophil count is low (< : (< 0.5/nl), various behavioural measures should be observed
In very rare cases, e.g. severe infections, the use of the haematopoietic growth factors G-CSF or GM-CSF to stimulate the body’s immune defence or the transfusion of white blood cell concentrates (granulocyte concentrates) may be considered.
You can find more detailed information in the Brochure „Infections? No thanks!” (German) by M. Exner, A. Simon and Deutsche Leukämie- & Lymphom-Hilfe.
Bleeding
In case of bleeding, platelet concentrates (PCs) can be transferred to avoid complications. As bleeding with low platelet counts can be life-threatening acute emergencies, immediate action must be taken in these cases. If the platelet count is very low, platelet concentrates can also be given as a preventive measure. The life span of platelets is only a few days. If there is very little or no production of platelets in the bone marrow, the administration of platelet concentrates may therefore be necessary several times a week.
Platelets carry tissue characteristics (HLA markers) that are different for each person. Some patients have antibodies against these HLA markers. This can happen spontaneously, during illness or after pregnancy. If such HLA antibodies are present, the transfused blood platelets are immediately destroyed and there is not a sufficient increase in platelets after a transfusion of a platelet concentrate. For these patients, special HLA-compatible platelet concentrates must be prepared from donors with matching HLA characteristics.
The use of platelet aggregation inhibitors such as acetylsalicylic acid (ASS) should be considered very carefully, particularly with very low platelet counts.
In women who have severe problems with menstrual bleeding, blood loss due to platelet deficiency can also be temporarily discontinued with hormone therapy, e.g. continuous administration of contraceptive pills or 3-monthly injections.
activities
BIn patients with aplastic anaemia, physical activity and exercise are recommended, depending on blood levels and the patient’s state of health. However, care should be taken to ensure that no excessive demands are made. It is therefore advisable to monitor the pulse rate during exercise. This is particularly important in anaemia, because if the red blood cell count is low, the body often tries to compensate for this deficiency by increasing the heart rate, which can lead to excessive strain on the heart. In the case of thrombocytopenia, sports with a risk of injury (e.g. martial arts or rock climbing), should be avoided at all costs.
Rehabilitation
If one’s participation in “normal” life can no longer be carried out as usual due to aplastic anaemia, rehabilitation measures, outpatient physiotherapy or psychological or psychotherapeutic care may be appropriate. These measures should be individually tailored to the patient.
If intensive therapy measures are planned, it makes sense to carry out rehabilitation measures only after these therapies. Physiotherapy or a psychological or psychotherapeutic care is also helpful to accompany the therapy.
If a child suffering from aplastic anaemia is reintegrated after treatment with immunosuppressive therapy or transplantation, a family-oriented measure in a paediatric-oncological aftercare facility can be useful due to the high psychosocial burden on the families.
The higher the granulocytecount and the lower the age of the patient at the time of diagnosis, the better the prognosis.
The data on survival after different therapies listed below are statistical data. This means that they cannot be automatically transferred to the individual patient. This list is only intended to give an overview of how possibilities and survival have improved in recent years. There are often subgroups that are not included here. For all specific forms of therapy, the results are significantly better for patients under 20 years of age than for patients over 20. The same applies to patients under 40 years of age compared to patients over 40 years of age. For stem cell transplants, the results are considerably better if the donor provides stem cells directly from the bone marrow and not peripheral peripheral stem cells obtained from the blood.
According to published data, overall survival in SAA/vSAA is after 3-6 years and separately for the different specific therapies:
Patients with evidence of a PNH clone can be entered in the international PNH Registry at the University Hospital in Essen in order to extend our knowledge about this group of AA patients.
If you are interested or have any questions, please email Prof Dr Alexander Röthalexander.roeth(at)uk-essen.de.
Patients with a telomeropathy can be included in thetelomeropathyregister at the Aachen University Hospital in order to gain further insights into this subgroup of AA patients.
If you are interested or have any questions, please email Prof Dr Tim H. Brümmendorftbruemmendorf(at)ukaachen.de.