
Hemoglobinuria paroxística nocturna (HPN)
1 ¿Qué es la HPN?
2 Síntomas
3 Diagnóstico
4 Evolución clínica
5 Tratamiento
6 Pronóstico
7 Deseo de tener hijos/embarazo
8 Registro
Se trata de una traducción automática. Nuestros textos no sustituyen la consulta con un médico. Si es posible, consulte siempre a uno de nuestros especialistas.
1 ¿Qué es la HPN?
1.1 Generalidades
La hemoglobinuria paroxística nocturna (HPN), al igual que la anemia aplásica, no es una enfermedad maligna, pero sí muy rara y potencialmente mortal. Se debe a un defecto genético adquirido de las células madre hematopoyéticas de la médula ósea, que no es hereditario.
1.2 Incidencia (epidemiología)
La frecuencia de la enfermedad (incidencia) es de 1-2 nuevos casos por cada millón de personas al año. Sin embargo, debido a la diversidad de los síntomas, se debe suponer que la HPN se diagnostica con poca frecuencia o demasiado tarde, ya que a menudo no se reconoce de inmediato. La enfermedad se diagnostica con mayor frecuencia entre los 25 y los 45 años, y afecta a ambos sexos con la misma frecuencia. No existe una acumulación familiar.
1.3 Origen (patogénesis)
La HPN se produce por una alteración genética (mutación) de las células madre hematopoyéticas de la médula ósea. Esta alteración no está presente desde el nacimiento, sino que se desarrolla a lo largo de la vida (mutación genética somática) y no se transmite a la descendencia. Las células sanas y enfermas coexisten (mosaico).
Esta mutación genética se encuentra típicamente en una sección específica del genoma, el gen PIG-A, y afecta a una o varias células madre hematopoyéticas de la médula ósea. El gen produce un biocatalizador (enzima) que normalmente se necesita para la producción de un sistema de anclaje especial, el anclaje glicosilfosfatidilinositol (anclaje GPI). Este se encuentra en la membrana celular y sirve para fijar numerosas proteínas en la membrana celular, que, entre otras cosas, regulan el sistema inmunológico. De esta manera, protegen a las células, por ejemplo, de un ataque por parte de una parte específica del sistema inmunológico, el llamado sistema del complemento, marcándolas como «no extrañas».
Dos de estas proteínas desempeñan un papel especialmente importante en este proceso:
- Factor acelerador de la desintegración del complemento (DAF, CD55)
- Protectina (MAC-IP: proteína inhibidora del complejo de ataque de membrana, MIRL: inhibidor de membrana de la lisis reactiva, CD59)
Una disminución o la ausencia total de proteínas ancladas a GPI en la membrana celular de los eritrocitos, leucocitos y trombocitos hace que estas células sean más vulnerables a la destrucción por parte del sistema del complemento. Esto provoca la rotura de los eritrocitos con HPN en los vasos sanguíneos (hemólisis intravascular) y a la activación de las plaquetas (PNH), lo que puede provocar trombosis.
El sistema del complemento sirve al organismo para defenderse de agentes infecciosos, parásitos, moléculas extrañas, etc. Cuando se activa, se inicia un proceso progresivo en cascada (cascada del complemento) que puede terminar, entre otras cosas, con la destrucción de la célula diana.
2 Síntomas
Los síntomas y molestias de la HPN pueden variar en intensidad y ser temporales o permanentes. Además, la activación del sistema del complemento por infecciones, embarazo o estrés puede provocar un aumento adicional de los síntomas, hasta llegar a una descomposición muy elevada de los glóbulos rojos (crisis hemolítica). En el caso de un tratamiento continuo con un inhibidor del complemento, se habla también de hemólisis de ruptura. Una señal de advertencia típica y visible de esto sería la aparición de orina oscura o negra (hemoglobinuria). Asimismo, el riesgo de trombosis aumenta durante este episodio.
En estas fases, a menudo se necesitan reservas de sangre (concentrados de eritrocitos, CE) u otras terapias.
Si la HPN no se trata, esta afección puede poner en peligro la vida.
2.1 Disminución del recuento celular (citopenias)
Anemia
La descomposición de los glóbulos rojos (hemólisis) puede provocar anemia y, por lo tanto, la falta de transportadores de oxígeno (hemoglobina).
Entre los síntomas se incluyen:
- Pálidez de la piel (signo inespecífico)
- Disminución del rendimiento, falta de concentración, depresión, fatiga, cansancio, pesadez en las piernas, fatiga rápida
- Dificultad para respirar (disnea) bajo esfuerzo como consecuencia de una disminución del número de transportadores de oxígeno
- Mareos, zumbidos en los oídos, aumento de la frecuencia cardíaca (taquicardia), opresión en el pecho (angina de pecho), trastornos visuales
La anemia puede ser tan grave que sea necesaria una transfusión de glóbulos rojos (concentrados de eritrocitos).
Otras series celulares reducidas
Además de la serie de glóbulos rojos, también pueden reducirse otras series de células sanguíneas (citopenia), como por ejemplo las plaquetas (trombocitopenia) o los granulocitos (neutropenia). Esto suele ser síntoma de anemia aplásica concomitante.
2.2 Efectos de la hemólisis
Cuando se descomponen los glóbulos rojos, la degradación de la hemoglobina provoca un aumento de la bilirrubina, un pigmento biliar, en la sangre. Esto puede provocar un color amarillento en la piel y en la parte blanca del globo ocular (esclerótica). En este caso se habla de ictericia (ictericia o ictericia escleral).
Además, la descomposición de los glóbulos rojos libera hemoglobina. Si la concentración de hemoglobina libre es muy alta, esta puede excretarse a través de los riñones y provocar una orina oscura, de color marrón rojizo o incluso negra (hemoglobinuria).
A través de diferentes etapas de degradación, la hemoglobina libre conduce a una menor disponibilidad de óxido nítrico (NO). El óxido nítrico es necesario para la relajación de la musculatura lisa, que se encuentra, por ejemplo, en el tracto gastrointestinal, en los vasos sanguíneos o en los pulmones. Si no hay suficiente disponibilidad, se produce una tensión de la musculatura lisa que provoca, entre otras cosas, espasmos, estrechamiento de los vasos sanguíneos y, por consiguiente, un aumento de la presión arterial.
Este mecanismo de acción explica muchos de los síntomas clínicos de la HPN:
- Dolores abdominales intensos, a menudo agudos
- Espasmos del esófago con dificultades para tragar (disfagia)
- Hipertensión arterial
- Presión alta en la circulación pulmonar (hipertensión pulmonar) con dificultad para respirar
- Deterioro de la función renal
- Disfunción eréctil (disfunción eréctil)
2.3 Fatiga
La fatiga es un cansancio o agotamiento anormalmente prolongado que puede afectar significativamente al rendimiento físico y mental. Se caracteriza por la ausencia de mejoría o una mejoría insuficiente, incluso con más descanso o sueño.
Una posible causa de la fatiga relacionada con la HPN, además de la anemia y la destrucción de los glóbulos rojos, es la consiguiente deficiencia de óxido nítrico (NO). Sin embargo, la fatiga también puede ser causada por otros trastornos del sistema inmunológico o del metabolismo.
2.4 Propensión a la trombosis (trombofilia)
Una de las consecuencias más peligrosas de la HPN no tratada y de la consiguiente falta de óxido nítrico (NO) es la formación de coágulos sanguíneos (trombosis). Esta deficiencia activa, entre otras cosas, las plaquetas, lo que provoca la formación de coágulos sanguíneos de forma antinatural. Estos coágulos pueden obstruir los vasos sanguíneos pequeños y provocar un infarto en los órganos afectados. Pueden verse afectadas diversas partes del cuerpo, por ejemplo, el hígado o el cerebro, tanto los vasos que transportan la sangre (venas) como los que la suministran (arterias). Del mismo modo, la elevada acumulación de hemoglobina libre puede provocar una insuficiencia renal aguda.
3 Diagnóstico
El método de exploración preferido para el diagnóstico de la HPN es la denominada citometría de flujo. Este método es muy sensible y, por lo tanto, puede detectar un número muy pequeño de células patológicamente alteradas. Además, permite determinar con gran precisión la proporción de células afectadas (tamaño del clon de HPN) y los tipos de células afectadas, por ejemplo, eritrocitos, granulocitos o monocitos. Para este examen se utiliza sangre de una vena (sangre periférica).
En el momento del diagnóstico inicial deben realizarse otros exámenes:
- Historial médico (anamnesis), también de la familia, incluyendo preguntas específicas sobre los síntomas típicos de la HPN, véase 2 Síntomas
- Exploración física teniendo en cuenta los aspectos especiales mencionados anteriormente: signos de anemia, ictericia, indicios de trombosis aguda o previa, signos de hemorragia, anomalías constitucionales como en las anemias aplásicas congénitas (véase allí), agrandamiento del bazo (esplenomegalia)
- Exámenes celulares
- Hemograma diferencial microscópico
- Examen de glóbulos rojos jóvenes e inmaduros en la sangre (reticulocitos)
- Química clínica
- Parámetros de hemólisis: en particular, LDH, haptoglobina, bilirrubina
- Parámetros de función renal: creatinina, saturación de transferrina
- Niveles de vitamina B12 y ácido fólico
- Estado del hierro: ferritina, transferrina, saturación de transferrina, hemoglobina reticulocitaria. Si los valores de ferritina son > 1000-2000 µg/l, dependiendo de la evolución, se debe realizar un examen para detectar posibles daños orgánicos causados por una sobrecarga de hierro.
- Valor de BNP en el suero sanguíneo para evaluar la función del ventrículo derecho como indicio de hipertensión pulmonar (hipertensión pulmonar)
- Diagnóstico funcional
-
- Ecografía abdominal (abdomen)
- Ecografía cardíaca
- Función pulmonar
- ECG
En el momento del diagnóstico inicial, se debe realizar un diagnóstico de la médula ósea con histología, citología, citogenética y genética molecular. Esto es especialmente importante si existe una citopenia tan grave que se sospecha que la HPN está relacionada con otra enfermedad hematológica, como anemia aplásica o síndrome mielodisplásico (SMD) .
Si se detectan células de HPN (clon de HPN) o se diagnostica un síndrome de insuficiencia medular (Bone Marrow Failure Syndrome, BMFS), deben realizarse controles cada 6 meses, especialmente durante los dos primeros años tras el diagnóstico inicial, así como cuando aparezcan nuevos síntomas. La proporción relativa del clon PNH (tamaño del clon PNH) en el número total de células de la médula ósea puede variar y es importante para el curso posterior de la enfermedad, así como para determinar y controlar el tratamiento.
4 Evolución clínica
4.1 Generalidades
Los síntomas y problemas de la HPN pueden ser muy variados y afectar a la calidad de vida debido a los síntomas mencionados anteriormente.
Los mecanismos descritos de la enfermedad pueden provocar, entre otras cosas, hipertensión arterial, hipertensión en la circulación pulmonar (hipertensión pulmonar) y disfunciones renales. Estos cambios pueden provocar daños permanentes y requieren un control constante de la evolución.
4.2 Coágulos sanguíneos (trombosis/tromboembolias)
La complicación más grave de la HPN es la formación de coágulos sanguíneos (trombosis/tromboembolias), que provocan la obstrucción de los vasos sanguíneos. Según los estudios, la probabilidad de que se produzca una trombosis es del
– del 30 % en un plazo de 10 años sin tratamiento específico (inhibidores del complemento, inhibidores de la agregación plaquetaria)
– del 30-50 % durante el curso de la enfermedad sin medidas terapéuticas específicas
Las complicaciones tromboembólicas son la causa de hasta el 67 % de todas las muertes en la HPN. Los pacientes con HPN que solo tienen unas pocas células HPN también tienen un mayor riesgo de trombosis. Es posible que la probabilidad de sufrir una trombosis dependa del tamaño del clon HPN.
Las trombosis en pacientes con HPN se producen en lugares típicos y menos típicos:

Las trombosis en pacientes con HPN se producen en lugares típicos y menos típicos.
4.3 Disfunción renal
Dos tercios de los pacientes con HPN presentan una alteración de la función renal. Predomina una alteración de la función de filtrado y, por lo tanto, un empeoramiento de la depuración del plasma sanguíneo por parte de los riñones. Como parámetro de la función renal se determina el denominado aclaramiento de creatinina , que indica el tiempo que tarda el plasma sanguíneo en eliminar la creatinina, un producto de degradación muscular. Tanto la función de filtrado como el aclaramiento de creatinina pueden empeorar a lo largo de la enfermedad.
4.4 Disminución del recuento celular (citopenias)
La disminución significativa de las células sanguíneas debido a una enfermedad concomitante de la médula ósea es, con un 20 %, la segunda causa más frecuente de complicaciones graves. Aproximadamente el 15 % de los pacientes desarrollan anemia aplásica con ausencia de las tres series de células sanguíneas (pancitopenia). Por el contrario, también puede presentarse primero una anemia aplásica, a la que luego se suma la HPN.
5 Tratamiento
5.1 Resumen
El tratamiento de la HPN depende de la gravedad de las molestias y los síntomas. Si no hay molestias relevantes, está justificado realizar solo controles frecuentes. Se pueden considerar medidas de apoyo y la inhibición farmacológica de la coagulación sanguínea (anticoagulación), también conocida como dilución de la sangre. La anticoagulación no diluye realmente la sangre, sino que evita la obstrucción («espesamiento») de los vasos sanguíneos por coágulos. En la HPN sintomática, el tipo de tratamiento depende, entre otras cosas, de la gravedad de la destrucción de los glóbulos rojos (hemólisis) y de la presencia de trombosis o coágulos sanguíneos.
En el siguiente gráfico se muestra el concepto de tratamiento de la HPN hemolítica:

Representación simplificada del algoritmo terapéutico. El algoritmo completo se muestra en las directrices de la DGHO en Onkopedia.
5.2 Terapia específica
Dado que el sistema del complemento Dado que el sistema del complemento (véase 4.1.3) ataca a los glóbulos rojos patológicos de la HPN, los medicamentos actúan en este punto. Se denominan inhibidores del complemento e inhiben una parte del sistema del complemento. De este modo, suprimen la destrucción de los glóbulos rojos o la evitan por completo. El objetivo de un tratamiento con un inhibidor del complemento de este tipo es evitar complicaciones graves y potencialmente mortales, así como posibles daños secundarios.
Las razones médicas para un tratamiento con eculizumab/ravulizumab son, entre otras, las siguientes:
- Coágulos sanguíneos
- Dolores abdominales agudos
- Crisis hemolíticas
- Alteración de la función renal relacionada con la HPN
- Necesidad de transfusiones
- Hipertensión pulmonar asociada a la HPN (hipertensión pulmonar)
Inhibición del complemento C5
Las terapias dirigidas contra la HPN autorizadas desde hace más tiempo son el anticuerpo anti-C5 eculizumab (Soliris®), introducido en 2007, y su forma de acción prolongada ravulizumab (Ultomiris®), autorizada en 2019.
Estos dos principios activos bloquean la actividad de la proteína C5 del sistema del complemento (lo que se denomina inhibición terminal del complemento). Diversos estudios han demostrado que, gracias a este bloqueo, el eculizumab y el ravulizumab reducen considerablemente la destrucción directa de los glóbulos rojos en los vasos sanguíneos (hemólisis intravascular).
Esto ha supuesto una mejora considerable de la calidad de vida de los pacientes con HPN tratados con eculizumab/ravulizumab. Esto se observó incluso en los pacientes en los que no se produjo una normalización de los niveles de hemoglobina. Esto sugiere que, además de la mejora de la anemia, parte del efecto puede atribuirse al bloqueo de la destrucción de los glóbulos rojos (hemólisis) en sí.
El tratamiento específico provocó una disminución significativa de la necesidad de transfusiones, la anemia, el agotamiento (fatiga), los dolores abdominales agudos, así como una reducción de la hipertensión arterial (hipertensión arterial) y la hipertensión pulmonar (hipertensión pulmonar). Además, en los estudios se describió una mejora o incluso una normalización de la función renal tras una posible disfunción renal previa.
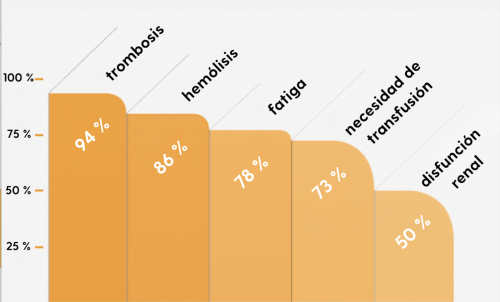
Efecto del eculizumab en el tratamiento de la HPN
El eculizumab o el ravulizumab se administran mediante infusión intravenosa (por vía intravenosa) y puede realizarse de forma ambulatoria. Por lo general, la infusión de eculizumab se administra una vez a la semana durante las primeras 4 semanas para alcanzar la saturación y, a continuación, cada 2 semanas como terapia de mantenimiento. Por el contrario, el ravulizumab debe administrarse dos veces con un intervalo de dos semanas para la saturación y, a continuación, solo cada ocho semanas en la terapia de mantenimiento. A diferencia del eculizumab, la dosis de ravulizumab depende del peso corporal. La duración de la infusión depende, a su vez, de la dosis.
Desde 2024, con el crovalimab (Piasky®) se dispone de otro anticuerpo anti-C5 que, tras una única administración intravenosa (infusión), puede administrarse bajo la piel (subcutáneamente) en un volumen menor. En este caso, es necesario un tratamiento de mantenimiento cada 4 semanas.
Mientras que la destrucción de los glóbulos rojos dentro de los vasos sanguíneos (hemólisis intravascular) se suprime con éxito mediante una inhibición terminal del complemento con eculizumab/ravulizumab/crovalimab, en algunos pacientes se produce ahora una lenta degradación de los glóbulos rojos patológicamente alterados fuera de los vasos (hemólisis extravascular). Este proceso se desencadena por una carga de precursores del sistema del complemento y puede detectarse mediante análisis de sangre especiales (entre otros, reticulocitos, nivel de bilirrubina, prueba de Coombs). La supresión de la destrucción de los glóbulos rojos fuera de los vasos sanguíneos fue el punto de partida para el desarrollo de nuevos medicamentos.
Inhibición del complemento C3
Los inhibidores del complemento C3 (inhibidores del complemento proximal) inhiben el complemento dentro de la cascada antes que los inhibidores C5. De este modo, se puede reducir o prevenir la carga de las células de la HPN con precursores del complemento y, con ello, la hemólisis extravascular. Entre estas nuevas sustancias se encuentran el inhibidor C3 pegcetacoplan, el inhibidor del factor D danicopan y el inhibidor del factor B iptacopan. Al igual que todo el sistema del complemento, los factores B y D son proteínas.
(Figura a continuación)
El pegcetacoplan (Aspaveli®) debe administrarse en forma de infusión 2-3 veces por semana durante aproximadamente 1 hora bajo la piel (subcutánea).
El danicopan (Voydeya®) en forma de comprimidos debe tomarse además de un tratamiento en curso con eculizumab o ravulizumab, en dosis de 150-200 mg 3 veces al día.
El iptacopán (Fabhalta®) se administra en forma de comprimidos (cápsulas) con 200 mg dos veces al día como monoterapia.
En general, los datos de los estudios de autorización y las evaluaciones actuales sobre el tratamiento a largo plazo muestran que se trata de terapias y medicamentos muy bien tolerados. Un efecto secundario muy frecuente es el dolor de cabeza, que solo se produce al inicio del tratamiento. Otros posibles efectos secundarios son, entre otros, infecciones, alteraciones en el hemograma, insomnio, molestias gastrointestinales, problemas cutáneos, síntomas similares a los de la gripe y fatiga.
Además, se observó que los pacientes con HPN se quejaban más de dolores de cabeza después de las primeras dosis de inhibición del complemento. Este síntoma es una señal de la buena eficacia del medicamento. Al descomponerse menos glóbulos rojos, vuelve a haber más óxido nítrico (NO) disponible. De este modo, los vasos sanguíneos pueden dilatarse, pero el cuerpo primero tiene que volver a acostumbrarse a este estado normal.
Al reducirse la descomposición de los glóbulos rojos enfermos, se excreta menos o nada de hemoglobina y, por lo tanto, nada de hierro a través de la orina. Al desaparecer esta pérdida crónica de hierro a través de los riñones, se observa un aumento de las reservas de hierro en algunos pacientes con HPN sometidos a tratamiento. Por lo tanto, estas deben controlarse regularmente, especialmente en caso de anemia aplásica relevante concomitante, para suspender a tiempo cualquier tratamiento en curso con comprimidos de hierro (ferritina objetivo > 100 µg/l) y, si es necesario, eliminar el exceso de hierro mediante la denominada terapia de quelación.
Sin embargo, el tratamiento con inhibidores del complemento no reduce el número de células enfermas ni cura la enfermedad. Por el contrario, , gracias a la protección de la inhibición del complemento, se descomponen menos o ninguno de los glóbulos rojos patológicamente alterados, que por lo tanto están presentes en mayor cantidad. El aumento de estos eritrocitos PNH o del nivel de hemoglobina es, por lo tanto, un signo de la eficacia del tratamiento. Por eso es tan importante administrar el tratamiento de forma regular o tomar los comprimidos con regularidad, para garantizar la protección continua de las células enfermas y evitar las complicaciones de la HPN mencionadas (hemólisis aguda).
Vacunas
Las vacunas preventivas son necesarias porque, debido a la inhibición del sistema del complemento, el organismo no puede defenderse adecuadamente contra las bacterias encapsuladas (meningococos, neumococos y Haemophilus influenzae). Estas bacterias pueden causar infecciones graves, como septicemia, neumonía o meningitis bacteriana. Por lo tanto, durante el tratamiento con eculizumab/ravulizumab/crovalimab es imprescindible vacunarse contra los meningococos. Es necesario cubrir el mayor número posible de cepas de meningococos (A, C, W135, Y al inicio del tratamiento y B durante el tratamiento) con dos vacunas diferentes (Menveo®/Nimenrix® y Bexsero®/Trumenba®), aunque no se alcanza una protección del 100 %. Las vacunas preventivas deben renovarse cada tres años.
Además, los pacientes tratados con pegcetacoplan o iptacopan deben vacunarse contra el neumococo (Prevenar 20®) y el Haemophilus influenzae tipo B (ActHib® o Hiberix®). La vacuna contra el neumococo debe repetirse cada 6 años. La vacuna contra el Haemophilus es única.
A pesar de todas las vacunas, pueden producirse infecciones graves con septicemia (sepsis), por lo que siempre deben seguirse estas normas de comportamiento:
En caso de fiebre (temperatura > 38,3 °C), erupción cutánea, malestar general intenso, rigidez en el cuello o infección respiratoria grave, es fundamental iniciar lo antes posible un «tratamiento de reserva» (por ejemplo, amoxicilina/ácido clavulánico 1000 mg) y acudir inmediatamente al médico para que realice más pruebas diagnósticas y, si es necesario, inicie un tratamiento antibiótico más amplio.
Además, se debe discutir con el hematólogo responsable el estado actual de vacunación, especialmente en lo que respecta a posibles vacunas adicionales, como la gripe, la COVID-19, el virus respiratorio sincitial (VRS), el herpes zóster y las vacunas de refuerzo.
5.3 Tratamiento sintomático
Además de los inhibidores del complemento mencionados, existen otras opciones para tratar los síntomas generales que se presentan.
En caso de síntomas de anemia, se pueden administrar concentrados de eritrocitos. Aunque la transfusión aporta pequeñas cantidades de factores del complemento, no se produce un aumento de la hemólisis mediada por el complemento. Esto también se aplica al uso de concentrados de trombocitos, véase Anemia aplásica, 5.5 Terapia de apoyo > Transfusiones de sangre.
Si la HPN no está directamente relacionada con una insuficiencia de la médula ósea, se denomina «HPN clásica». Si no se trata, a menudo se produce una deficiencia de hierro debido a la pérdida constante de hemoglobina a través de los riñones. Dado que el hierro es necesario para la formación de glóbulos rojos, en estas situaciones es necesario administrarlo. El hierro se puede administrar en forma de comprimidos o mediante infusión intravenosa. Lo ideal es tomar los preparados orales cada dos días por la mañana con el estómago vacío. Además, el hierro no debe tomarse al mismo tiempo que antibióticos o medicamentos para neutralizar la acidez estomacal (antiácidos). Una vez iniciado el tratamiento con hierro, la decisión terapéutica sobre la suplementación con hierro debe revisarse periódicamente. Para ello, es conveniente controlar los niveles de ferritina en el marco de los chequeos médicos (ferritina objetivo > 100 µg/l).
Debido al aumento compensatorio de la formación de glóbulos rojos, existe una mayor necesidad de ácido fólico y, en algunos casos, también de vitamina B12. En general, se debe tomar un suplemento de, por ejemplo, 5 mg de ácido fólico al día o 1-2 veces por semana. En caso de deficiencia, la vitamina B12 debe administrarse como tratamiento durante un periodo de tiempo determinado.
En estudios anteriores se observó que el riesgo de trombosis está relacionado con la cantidad de células PNH y la intensidad de la actividad hemolítica. Si la proporción de granulocitos con alteración de la GPI es superior al 50 % y o el valor de LDH es más de 1,5 veces superior al valor normal superior, la incidencia de trombosis aumenta significativamente. Cuando se administró un anticoagulante preventivo a estos pacientes, desarrollaron trombosis con mucha menos frecuencia.
Por lo tanto, se aplican las siguientes recomendaciones:
- El uso de un medicamento anticoagulante debe decidirse de forma individual para cada paciente.
- No es necesario administrar anticoagulantes preventivos durante el tratamiento con un inhibidor del complemento.
- Si se ha iniciado un tratamiento anticoagulante preventivo antes de la terapia con un inhibidor del complemento, se puede considerar la posibilidad de interrumpirlo una vez que se haya normalizado la actividad hemolítica bajo la terapia con un inhibidor del complemento. Sin embargo, se desaconseja encarecidamente suspenderlo por cuenta propia.
- Si ya se ha producido una trombosis, se debe realizar una anticoagulación. La duración depende de la localización de la trombosis y de su evolución.
- En situaciones de riesgo, como por ejemplo, inmovilización en cama, restricción prolongada del movimiento (yeso), operaciones o viajes largos (> aprox. 4-6 horas) en autobús o avión, se debe realizar una anticoagulación preventiva (por ejemplo, en forma de inyección o comprimido) en función de los valores plaquetarios actuales.
- Las infecciones bacterianas deben detectarse a tiempo y tratarse de forma sistemática con antibióticos, ya que las infecciones pueden provocar un empeoramiento agudo de la HPN con una crisis hemolítica.
- En caso de crisis hemolítica/hemólisis aguda, se debe garantizar una hidratación suficiente. En caso necesario, se requerirá un tratamiento con antibióticos, transfusiones y dosis adicionales del inhibidor del complemento, así como diálisis en caso de deterioro de la función renal.
- En casos excepcionales, si se presenta insuficiencia medular (insuficiencia de la médula ósea) o se desarrolla anemia renal, puede ser conveniente administrar factores de crecimiento hematopoyéticos, como eritropoyetina o G-CSF.
- Si la insuficiencia de la médula ósea (aplasia) es más importante que la HPN, se debe realizar un tratamiento inmunosupresor o un trasplante de células madre o de médula ósea, véase Anemia aplásica, 5 Tratamiento.
5.4 Curación
El único tratamiento de la HPN con perspectivas de curación (enfoque curativo) es el trasplante alogénico de médula ósea o células madre. Sin embargo, conlleva una tasa considerable de complicaciones (morbilidad) y mortalidad. Por lo tanto, la decisión terapéutica (indicación) para un trasplante debe tomarse con mucho rigor, especialmente desde que se dispone de la terapia con inhibidores del complemento.
Las razones médicas para un trasplante de células madre son
- Complicaciones tromboembólicas recurrentes y potencialmente mortales que no responden a otros tratamientos.
- Anemia hemolítica muy grave (refractaria) que no responde al tratamiento y requiere transfusiones.
- Presencia de un clon de HPN en una anemia aplásica (AA) o un síndrome mielodisplásico (SMD), cuando la anemia aplásica o el síndrome mielodisplásico ya constituyen un motivo para el trasplante
- Transición a anemia aplásica, cuando no está indicada una terapia inmunosupresora, o a síndrome mielodisplásico
6 Pronóstico
Gracias a la notable reducción de los eventos tromboembólicos con el tratamiento con un inhibidor del complemento, los pacientes diagnosticados con HPN tienen prácticamente la misma esperanza de vida que la población normal.
7 Deseo de tener hijos/embarazo
Hasta hace pocos años, se desaconsejaba a las pacientes con HPN quedarse embarazadas, ya que a menudo se observaban complicaciones potencialmente mortales tanto para la madre como para el niño. Dado que este riesgo se ha reducido considerablemente con la terapia con anticuerpos, la planificación familiar vuelve a cobrar importancia. Entretanto, se dispone de informes sobre embarazos durante el tratamiento con eculizumab, ravulizumab y crovalimab, que muestran resultados muy alentadores, aunque el número de casos es limitado. Los embarazos transcurrieron sin complicaciones y todos los niños nacieron sanos. No obstante, si se desea tener hijos o se está embarazada, se debe acudir a un centro especializado con departamentos de hematología y ginecología para aclarar el perfil de riesgo individual de la paciente y, si es necesario, ajustar la dosis del inhibidor del complemento.
8 Perspectivas
Actualmente se están probando en ensayos clínicos otras sustancias que inhiben el sistema del complemento.
9 Registro
Dado que la HPN es una enfermedad muy rara, la información relevante sobre la enfermedad y su tratamiento solo puede obtenerse mediante la evaluación de los datos del mayor número posible de pacientes con HPN a nivel internacional. Con este fin, se creó inicialmente el Registro Internacional de Pacientes con HPN (PNH Registry), que se cerró a finales de 2024. Para continuar con esta actividad, especialmente con las nuevas opciones terapéuticas, un grupo internacional de expertos en HPN (International PNH Interest Group, IPIG) creó un nuevo registro. Desde 2025, tras obtener el consentimiento de los pacientes, documenta cada seis meses, de forma anónima, datos sobre la evolución de la enfermedad, el tratamiento y la calidad de vida. Dado que solo a través de esta información se pueden obtener nuevos conocimientos sobre la enfermedad y lograr una mejora adicional del tratamiento, todos los pacientes con HPN deberían estar dispuestos a facilitar sus datos al registro.
¡PARTICIPE!
Si está interesado o tiene alguna pregunta, póngase en contacto con el Prof. Dr. med. Alexander Röth (alexander.roeth(at)ukessen.de) o al Prof. Dr. med. Hubert Schrezenmeier (h.schrezenmeier(at)blutspende.de).