
Paroxysmal Nocturnal Haemoglobinuria (PNH)
1 What is PNH?
- Complement Decay Accelerating Factor (DAF, CD55)
- Protectin (MAC-IP: Membrane Attack Complex Inhibitory Protein, MIRL: Membrane Inhibitor of Reactive Lysis, CD59)
1.1 General
Paroxysmal nocturnal haemoglobinuria (PNH), like aplastic anaemia (AA), is not malignant, but is a very rare, acquired and life-threatening disease. It is due to a defect in the blood-developing stem cells of the bone marrow, and is not inherited.
1.2 Occurrence (epidemiology)
The disease frequency (incidence) is 1-2 occurrences per million people per year. However, due to the diversity of its symptoms, PNH is rarely recognised and is therefore underdiagnosed. The disease is usually diagnosed in people between the ages of 25 and 45. The frequency of diagnosis is about the same for men and women. There is no familial predisposition.
1.3 Origin (pathogenesis)
PNH is caused by a mutation (genetic modification) of blood-developing stem cells in the bone marrow. This modification is not present from birth, but only occurs in later life somatic gene mutation and cannot be passed on to children. Healthy and sick cells can be present together at the same time (mosaic).
This gene mutation is typically found in a particular section of the genome, the PIG-a gene, and affects one or more types of blood-developing stem cells in the bone marrow. The gene produces an enzyme (a biological catalyst) that is normally required for production of a special anchor system, the glycosylphosphatidylinositol anchor GPI-anchor). This is located on the cell membrane and serves to attach numerous proteins to the cell membrane; these proteins are involved in regulation of the immune system. In this way, they protect the cells from an attack by a certain part of the immune system, the ”complement system’, by marking the cells as ‘self’ or non-foreign.
Two of these proteins play a particularly important role:
Loss or complete absence of GPI-anchored proteins on the cell membrane of erythrocytes, leukocytes and platelets makes these cells more susceptible to destruction by the complement system. This leads to a rupture of the erythrocytes in the blood vessels (intravascular haemolysis) and activation of the platelets, which can lead to thrombosis.
The complement system serves to defend the body against infectious agents, parasites, foreign molecules, etc. Once activated, a progressive cascade-like process (complement cascade) starts, which can end with destruction of the target cell.
- Pale skin (non-specific sign)
- Loss of stamina, lack of concentration, depression, fatigue, heavy legs and rapid fatigue
- Shortness of breath (dyspnoea) under load due to a smaller number of oxygen carriers
- Dizziness, ringing in the ears, increased heart rate (tachycardia), chest pain (angina pectoris), visual disturbances
- Severe, often crisis-type abdominal pain
- Cramping of the oesophagus with swallowing disorders (dysphagia)
- High blood pressure (Hypertension)
- High blood pressure in the lung circulation (pulmonary hypertension) with shortness of breath Renal insufficiency Erectile dysfunction
- Renal insufficiency
- Erectile dysfunction
2.1 Low cell counts (Cytopenia)
Anaemia
Destruction of red blood cells (haemolysis) can lead to anaemia and the loss of oxygen carriers (haemoglobin).
Symptoms include:
Anaemia may be so severe that transfusion of red blood cells (erythrocyte concentrates) becomes necessary.
Further loss of cell lines
In addition to the red blood cell line, other blood cell lines may be reduced (cytopenia), such as platelets (thrombocytopenia) or granulocytes (neutropenia).
2.2 Effects of haemolysis
Destruction of red blood cells leads to increased levels of bilirubin (a bile pigment) in the blood. This may cause the skin and the sclera (white outer layer of the eyeball) to turn yellow. This is also referred to as jaundice.
Haemoglobin is also released when the red blood cells break down. If the levels of free haemoglobin become excessive, some can be eliminated via the kidney, resulting in dark, red-brown urine (haemoglobinuria).
Free haemoglobin leads to reduced availablity of nitric oxide (NO) via various intermediate steps. Nitric oxide is required for relaxation of smooth muscle (located e.g. in the gastrointestinal tract and lungs); insufficient levels of NO result in increased tension of the smooth muscles, e.g. spasms and constriction of vessels.
This explains many of the clinical symptoms of PNH:
2.3 Fatigue
Fatigue is unusually persistant tiredness or exhaustion that may significantly impair physical and mental performance. It is characterised by the fact that increased rest or sleep results in little or no improvement.
Potential causes of fatigue are anaemia and destruction of red blood cells with the resulting lack of nitric oxide (NO), as well as disorders of the immune system and/or metabolism.
2.4 Thrombosis tendency (Thrombophilia)
One of the most dangerous consequences of the lack of nitric oxide is formation of blood clots (thrombosis). The platelets are presumably activated by the NO deficiency, and this causes abnormal clots. These can occur in various parts of the body, including the liver or brain and both arteries and veins. The symptoms and complaints associated with PNH may be permanent. In addition, activation of the complement system by infections, pregnancy or stress may lead to further exacerbation of the symptoms, including greatly increased destruction of red blood cells (haemolytic crisis). This condition may be life-threatening if not treated. Blood clots can clog the small blood vessels in the kidneys, which can lead to acute kidney failure. There is also an increased risk of thrombosis during these haemolytic crises. In these situations packed RBCs (erythrocyte concentrates) are often required.
- The patient’s (and family’s) medical history (anamnesis) are taken, including a targeted survey of symptoms typical of PNH (see Section 2 Symptoms
- Physical examination with regard to the special aspects mentioned above: signs of anaemia or jaundice, indications of acute or previous thrombosis, evidence of bleeding, constitutional abnormalities such as congenital aplastic anaemia (see Chapter 3 AA), and enlargement of the spleen (splenomegaly)
- Cell investigations
-
-
- Microscopic differential haemogram
- Reticulocytes
-
-
- Clinical chemistry
-
-
- Haemolysis parameters: in particular LDH, haptoglobin, bilirubin
- Renal function parameters: creatinine, urea
- Levels of vitamin B12 and folic acid
- Iron status: ferritin, transferrin, transferrin saturation, reticulocyte haemoglobin. With ferritin values> > 1000 µg/l, further clarification of possible organ damage due to possible iron overload
- BNP (B-type natriueretic peptide) value in the blood serum to assess function of the right heart
-
-
- Functional diagnostics
-
-
- Ultrasound of abdomen
- Lung function
- ECG
-
-
The method used to diagnose PNH is flow cytometry. This method is very sensitive and can detect a very small number of cells that have been altered as a result of the disease. In addition, the proportion of affected cells (the PNH clone size) and the affected cell types (e.g. erythrocytes or granulocytes) are determined very accurately. Venous (peripheral blood) is used for this test.
Additional tests are usually carried out during the initial diagnosis:
Diagnostic tests of bone marrow with cytology, cytogenetics, and histology should be performed at the time of the initial diagnosis. This is particularly important if there is also cytopenia severe enough that PNH in connection with another haematological disease (e.g. aplastic anaemia or myelodysplastic syndrome (MDS)) must be suspected. With evidence of PNH cells (PNH clone) or diagnosis of a bone marrow insufficiency syndrome, check-ups should be carried out every 6 months, especially in the first two years after initial diagnosis and in the case of new symptoms. It is important to measure the extent of the PNH clone as a proportion of the total number of bone marrow cells. This parameter is useful for estimating the prognosis and managing treatment of the disease.
4.1 General
The symptoms and handicaps associated with PNH can vary widely and may lead to deterioration of the quality of life.
Due to the mechanisms of the disease (mentioned above), arterial and pulmonary hypertension (high blood pressure in the body and lungs) as well as kidney dysfunction can occur. These problems can lead to permanent damage and require continuous follow-up.
4.2 Blood Clots (Thrombosis/Thromboembolism)
The most feared complication of PNH is formation of blood clots (thrombosis/thromboembolism), which can block blood vessels. According to trials, the probability of thrombosis occurring without specific PNH therapy was over 30% within 10 years. Approximately 30-50% of all PNH patients develop thrombosis in the course of their disease without specific treatment measures. Complications of thromboembolism are responsible for up to 67% of all deaths due to PNH. The probability of thrombosis may also depend on the number of PNH cells, although even patients with only a few PNH cells have an increased risk of thrombosis.
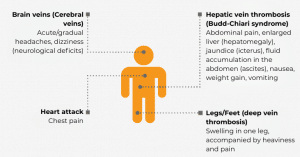
4.3 Kidney disease
Two-thirds of all patients with PNH have kidney disease. This is usually due to impairment of the filtering function of the kidney, and therefore its ability to purify the blood plasma. The parameter for plasma purification is creatinine clearance, which is a measure of how long it takes for creatinine (a product of muscle degradation) to be cleared from the blood plasma. The filter function and creatinine clearance may both deteriorate during the course of the disease.
4.4 Reduced cell numbers (Cytopenias)
Significant diminution of blood cell numbers due to concomitant bone marrow disease is the second-most common cause of serious complications (20%). In the course of PNH, approximately 15% of patients develop aplastic anaemia with the absence of all three blood cell lines (pancytopenia). Conversely, aplastic anaemia can develop first and be followed by PNH.
- Blood clots
- Crisis-type abdominal pain
- Haemolytic crises
- PNH-related impairment of kidney function
- Need for transfusion
- Pulmonary hypertension associated with PNH
-
- The use of anticoagulant medication should be decided on an individual basis for each patient.
- Prophylactic clotting inhibitors are not necessary with eculizumab therapy.
- If prophylactic clotting inhibition has been initiated prior to eculizumab therapy, one should consider discontinuing it once haemolytic activity has normalised during eculizumab therapy. However, patients should under no circumstances discontinue treatment without consulting their doctor. However, patients should under no circumstances discontinue treatment without consulting their doctor.
- Anticoagulation should be applied if thrombosis has already occurred. The duration depends on the location of the thrombosis and its clinical course.
- In the case of risk situations such as being bed-bound, long-term restriction of movement (plaster cast), surgery or long trips > (> 4 hours in a bus or aeroplane), clotting inhibitors should be taken as a prophylactic measure according to the current platelet values.
- Bacterial infections need to be detected at an early stage and treated appropriately with an antibiotic, since infection can lead to acute deterioration of PNH and even a haemolytic crisis.
- Adequate fluid intake (hydration) should be ensured in the event of a haemolytic crisis. Anti-infection therapy, transfusions and eculizumab may be required; if the kidneys are endangered, purification (dialysis) may be necessary.
- In exceptional cases of concomitant bone marrow disorder (bone marrow insufficiency) or after the development of kidney-related (renal) anaemia, administration of haematopoietic (blood-forming) growth factors, e.g. erythropoietin or G-CSF, may be useful.
- If bone marrow deficiency (aplasia) is a greater concern than PNH, immunosuppressive therapy, stem cell or bone marrow transplantation should be performed (see .3.5 Aplastic Anaemia, 5 Treatment.
-
-
- Recurrent, life-threatening thromboembolic complications that do not respond to any other therapy
- Very severe (refractory) haemolytic anaemia that is not affected by therapy and requires transfusions
- Presence of PNH in addition to aplastic anaemia (AA) or myelodysplastic syndrome (MDS), if AA or MDS already justify a transplant
- Transition to aplastic anaemia or myelodysplastic syndrome
5.1 Overview
How PNH is treated depends on the patient’s symptoms. In the absence of relevant symptoms, only detailed check-ups are justified. Supportive measures and medication to inhibit blood clotting (anticoagulation), incorrectly referred to as ‘blood thinning’, may be considered. With symptomatic PNH, the type of treatment depends, among other things, on the severity of blood cell destruction (haemolysis) and the presence of blood clots.
The following diagram illustrates the treatment concept used for haemolytic PNH.

Simplified presentation of the therapy algorithm. For the complete algorithm, please visit Onkopedia (German).
5.2 Specific treatment
Since the complement system attacks the diseased red blood cells, partial inhibition of the system can suppress such destruction.
Since 2007, the only treatment available for PNH therapy has been the eculizumab antibody, which must be infused every two weeks. In July 2019, ravulizumab (a development of eculizumab) was authorised in the EU. The new antibody is administered as an infusion every eight weeks, and according to one trial it is not less effective than and and compatble with the previous standard therapy.
The following description relates to eculizumab, for which data is available for a much longer timespan. The information on function, precautions and behavioural recommendations is analogous to that for treatment with ravulizumab.
Eculizumab blocks activity of the C5 protein, part of the complement system. Various studies have shown that with this blockade, eculizumab greatly reduces the destruction of red blood cells in the blood vessels (intravascular haemolysis). There was a significant improvement in the quality of life for PNH patients treated with eculizumab. This improvement was even observed in patients who did not experience normalisation of haemoglobin levels. This suggests that, in addition to improving anaemia, part of the effect is due to blockage of the blood destruction (haemolysis) itself. As a further consequence of the reduced destruction of the red blood cells, there was a significant decrease in the transfusion requirement, anaemia, fatigue, crisis-type abdominal pain, as well as a reduction in high blood pressure (arterial hypertension) and pulmonary hypertension. In addition, clinical trials showed improvement or even normalisation of kidney function after a previous renal impairment.
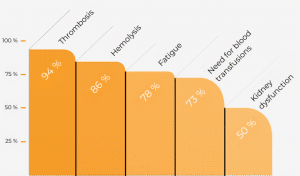
Medical reasons for treatment with eculizumab include
The aim of treatment is to prevent serious, life-threatening complications and resulting damage to the affected organs. Eculizumab is administered as an infusion into a vein (intravenous) and can be administered on an outpatient basis. The infusion is usually given once a week for saturation in the first four weeks, and then every two weeks as maintenance therapy. The data from authorisation studies and current evaluations of long-term therapy show that this therapy is very well tolerated. Headaches are a very common side effect at the start of therapy. Infections, changes in blood count, insomnia, gastrointestinal symptoms, skin problems, flu-like symptoms and fatigue are among the common side effects. Due to inhibition of the complement system, the body cannot provide sufficient protection against certain bacteria (meningococci), which can cause blood poisoning or bacterial meningitis. Vaccination against meningococcal disease is therefore absolutely necessary during treatment with eculizumab. Two different vaccines are recommended (Menveo®/Nimenrix® and Bexsero®/Trumenba®) to cover as many of the meningococcal strains as possible (a, C, W135, Y at the start of therapy and B during ongoing therapy), although 100% protection is not achieved. Booster vaccinations should be given every three years.
In case of fever ( > 38,0 °C), rash, strong feeling of illness or neck stiffness, it is crucial that “stand-by therapy” (e.g. ciprofloxacin 750 mg) is provided as soon as possible and a doctor is consulted immediately to initiate further diagnosis and, if necessary, extended antibiotic therapy. If ciprofloxacin cannot be given, amoxicillin or clavulanic acid 1000 mg are stand-by options.
Clinical trials showed that PNH patients complained more of headaches after the initial doses of eculizumab. This is a sign of the effectiveness of the medication. By decreasing the destruction of the red blood cells, more nitric oxide (NO) is available and the blood vessels can expand, but the body needs to get used to this normal condition. However, eculizumab therapy does not reduce the number of damaged cells or cure the disease. On the contrary, the antibody protection means that fewer of the diseased red blood cells disintegrate, so that more of them remain. An increase in the number of these erythrocytes is therefore a sign of effective therapy. Adherence to regular treatment intervals (every 14 +/- 2 days) is essential to ensure continuous protection of the injured cells and to avoid the complications of PNH (breakthrough haemolysis). Due to the reduced destruction of the damaged red blood cells, little or no haemoglobin (and thus iron) is excreted in the urine. Due to the elimination of this chronic loss of iron via the kidney, increased iron storage is observed in some PNH patients receiving eculizumab therapy. These patients should therefore be monitored regularly (especially if they have concomitant aplastic anaemia), to ensure that they discontinue any ongoing treatment with iron tablets at an early stage and, if necessary, that they remove excess iron with chelate therapy. Despite the successful suppression of cell destruction in the blood vessels (intravascular haemolysis) by eculizumab, a minor degradation of the diseased red blood cells does take place outside the vessels . (extravascular haemolysis). This can be detected with a special blood test (Coombs test)
5.3 Symptomatic treatment
In addition to specific treatment of PNH with eculizumab, there are other options for treating the symptoms. Symptoms of anaemia can be treated with packed RBCs. Although small amounts of complement factors are added by transfusion, there is no increase in complement-mediated haemolysis. This also applies to the use of platelet concentrates, see Chapter Aplastic Anaemia, 5.5 Supportive therapy > Blood transfusions.
If affection with PNH is not directly related to bone marrow failure, it is referred to as “classical PNH”. If PNH is not treated, iron deficiency often develops due to the ongoing loss of haemoglobin via the kidneys. As iron is necessary for the development of red blood cells, it must be supplemented in such situations. Iron can be administered in the form of tablets or a venous infusion. Oral preparations should be taken daily or every other day on an empty stomach. Iron should not be taken simultaneously with antibiotics or medications for neutralising stomach acid (antacids). If appropriate iron treatment is started, the decision to take iron supplements must be reviewed regularly. For this purpose, ferritin levels should be monitored as part of the regular medical check-ups. Due to the increased (compensatory) development of red blood cells, there is an increased need for folic acid and possibly vitamin B12. Supplementation should generally be in the form of e.g. 5 mg folic acid per day. Vitamin B12 should be taken according to current levels. In previous studies, it was found that the risk of thrombosis is associated with the quantity of PNH cells and the severity of haemolytic activity. The occurrence of thrombosis increases significantly if the proportion of GPI-deficient granulocytes is more than 50% and/or the LDH value is more than 1.5 times the upper limit. If such patients are given prophylactic clotting inhibitors, they develop significantly less thrombosis.
The following recommendations therefore apply:
In addition, the current vaccination status should be discussed with the treating haematologist, especially with regard to vaccination against pneumococcal pneumonia and influenza viruses.
5.4 Healing
The only treatment of PNH with the prospect of a cure (curative approach) is transplantation of allogeneic bone marrow or stem cells. However, this is accompanied by a significant rate of complications (morbidity) and mortality. Therefore, the indication (therapeutic decision) for a transplant should be considered very carefully, especially since antibody therapy with eculizumab is now an option.
Medical reasons for a stem cell transplant are:
5.5 Prospects
Several new substances that inhibit the complement system are currently being investigated inClinical trials. Ravulizumab which received admission in the EU on 2 July 2019 is being tested for subcutaneous application in clinical trials.
PNH patients now have approximately the same life expectancy as the normal population, since eculizumab therapy significantly reduces the occurrence of thromboembolic events.
Until a few years ago, PNH patients were advised not to become pregnant, because life-threatening complications were often observed in the mother and child. This risk has been significantly reduced with antibody therapy, so the issue of family planning arises again. Meanwhile, reports of pregnancies during eculizumab treatment are available and show very encouraging results, although the number of cases is limited. These pregnancies were basically without complications, and all the children have been healthy. However, if women wish to become (or already are) pregnant, they should seek support from a specialised centre with haematological and gynaecological expertise in order to clarify the individual risk profile of the patient and, if necessary, to adjust the dose of eculizumab.
Since PNH is an extremely rare disease, relevant information on the disease and its treatment can only be obtained by analysing the data of as many PNH patients as possible at an international level.
The International PNH Patient Registry was established for this purpose. It documents data on the course of the disease and the quality of life in anonymised form every six months after receiving patient approval. Since new knowledge of the disease and further improvement of the therapy can only be achieved on the basis of such information, as many PNH patients as possible should make their data available to the Registry.
PLEASE HELP!
If you would like to help or have any questions, please email Prof Dr Alexander Röth (alexander.roeth(at)ukessen.de) or Prof Dr Hubert Schrezenmeier (h.schrezenmeier(at)blutspende.de).